
CASO 119
13 de diciembre
Dr. Reinhard Rodríguez y Dra. Vanessa García Valencia. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Hombre, 30 años, múltiples pápulas asintomáticas, color piel, de 0,4 cc en el pecho. Sin antecedentes familiares.
¿Cuál cree usted es el diagnóstico más probable?
A. Esteatocistoma múltiple
B. Nevus del folículo piloso
C. Siringomas
D. Quiste velloso eruptivo
Solución y cierre del caso 23 de diciembre
CASO 112 (1ra y 2da parte)
3 de Mayo
Dra. Camila Montoya. Patóloga-Dermatopatóloga. Asunción, Paraguay. Dr. Rodrigo Restrepo. Patólogo-Dermatopatólogo. Profesor Programa de Especialización en Dermatopatología. Universidad CES. Medellín, Colombia.
Caso cortesía Dra. Ana María Aristizabal. Dermatológa. Profesora Univerdidad CES, Mdlln, Colombia.
Niña, 13 años. Áreas alopécicas fronto temporo parietales. Se tomaron dos grupos de biopsias en momentos diferentes (F1 primero; F2, F3 y F4 meses después)
Con los hallazgos histológicos ¿cuál cree es el diagnóstico más probable?
A. Alopecia androgénica
B. Alopecia frontal fibrosante
C. Alopecia areata en patrón ofiásico
D. Alopecia por tracción
Fotos clínicas 8 de mayo
Solución y cierre del caso 18 de mayo
 F1 |  F2 |  F3 |
|---|---|---|
 F4 |  F5 |
CASO 120
18 de enero
Dr. Reinhard Rodríguez y Dr. Javier Hernández. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Caso y fotos clínicas cortesía Dra. Carolina Velásquez. Profesora Dermatología Universidad CES. Mdlln, Col
Paciente masculino de 85 años quien presenta mácula hiperpigmentada en la frente.
¿Cuál cree usted es el diagnóstico más probable?
A. Queratosis actínica pigmentada
B. Lentigo solar
C. Nevus displásico
D. Lentigo maligno
Solución y cierre del caso 30 de enero
CASO 119
13 de diciembre
Dr. Reinhard Rodríguez y Dra. Vanessa García Valencia. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Hombre, 30 años, múltiples pápulas asintomáticas, color piel, de 0,4 cc en el pecho. Sin antecedentes familiares.
¿Cuál cree usted es el diagnóstico más probable?
A. Esteatocistoma múltiple
B. Nevus del folículo piloso
C. Siringomas
D. Quiste velloso eruptivo
Solución y cierre del caso 23 de diciembre
Si no te has inscrito, para opinar debes unirte a nuestra lista de correo AQUí
ESTACIÓN PIEL
Programa de Especialización en Dermatopatología
Universidad CES
Casos 2018 Julio a Diciembre
CASO 89
30 de noviembre
Dra. Ana Sanín. Residente de Dermatología. CES. Medellín , Colombia
Dra. Ana Londoño. Residente de Dermatología. UPB. Medellín , Colombia
Dra. Camila Montoya. Dermatopatóloga. Asunción. Paraguay
Dr. Andrés Floréz. Jefe Programa de Especialización en Dermatopatología. Universidad CES. Medellín , Colombia
Mujer, 44 años de edad. Pápulas purpúricas de un año de evolución localizadas en labio superior e inferior, las cuales son asintomáticas.
Respuestas de los participantes comienzan a aparecer el 10 diciembre . Solución y cierre de caso 15 de diciembre 2018
 F1.Nódulo violaceo |  F2Panorámica. En dermis vaso venoso dilatado |  F3Vaso venoso dilatado y trombosado |
|---|---|---|
 F4Mayor detalle. Vaso venoso dilatado y trombosado |  F5Mayor detalle. Vaso venoso dilatado y trombosado |
Con las características histológicas observadas, el diagnóstico más probable es:
A. Nevus azul.
B. Lago venoso.
C. Mucocele.
D. Pigmentación no melánica por depósitos exógenos
Respuestas
Lago venoso María del Carmen Gómez
Lago venoso Guillermo Ramos
Lago venoso Beatriz di Martino
Lago venoso Juan M. González
Lago venoso Soledad Machado
Lago venoso Juan David Ruiz
Lago venoso Sandra Quijano M.
Lago venoso Andrés F. López
Lago venoso Fernando Cabo G.
Lago venoso Juliana Calle
Lago venoso Griselda de Anda
Lago venoso Oryana P. Meza
Lago venoso Jaime Arturo Mejía
Lago venoso María del Valle Centeno
Lago venoso Víctor Delgado
Lago venoso Gustavo Camino
Lago venoso Ramiro Pinedo
Lago venoso María Janeth Vargas
Lago venoso Regina Barros Domínguez
Lago venoso Ismery Cabello
Lago venoso Fredy Salazar
Lago venoso Clara Jaramillo
Lago venoso Carolina Cabrera Salom
Lago Venoso Mary Miller
Respuesta:
B- Lago Venoso (correcta)
Es la segunda patología benigna mas frecuente de los labios, luego del granuloma telangiectásico. Clínicamente son pápulas o nódulos violáceos usualmente asintomáticos localizados principalmente en el bermellón (F1). Histológicamente son una ectasia vascular formada por vénulas dilatadas, pudiéndose desarrollar trombos(F2-5); estos cambios se dan por pérdida de rigidez de los tejidos asociado a un aumento de la presión venosa (3,4).
A-Nevus azul (incorrecto): Más frecuentes en mujeres. También se presentan como pápulas o nódulos, pueden ser pigmentados. Afecta principalmente el paladar blando, y con menos frecuencia bermellón. Histológicamente son muy diferentes, se ven células melanocíticas dendríticas fusiformes, altamente pigmentadas (4).
C- Mucocele (incorrecto): El mucocele oral es la lesión benigna más común de la glándula salival menor. Se presenta como uno o múltiples nódulos fluctuantes y suaves eritematosos o azules, principalmente en el labio inferior. A la histología se observan pequeños espacios quísticos con mucina y células llenas de mucifagos. (1).
D- pigmentación no melánica por depósitos exógenos (incorrecta): Incluyen el tatuaje por amalgamas dentales y la pigmentación por metales pesados. Son lesiones pequeñas 0,1 a 2,0 cm. Histológicamente se observan partículas de amalgama a lo largo de las fibras de colágeno y alrededor de los vasos sanguíneos.(2).
Bibliografia
1. More CB, et al. Oral mucocele: A clinical and histopathological study. J Oral Maxillofac Pathol;2014;18(Suppl 1):S72-77. PubMed Freetextlink
2. Meleti M, et al. Pigmented lesions of the oral mucosa and perioral tissues: a flow-chart for the diagnosis and some recommendations for the management. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. mayo de 2008;105(5):606-16. PubMed
3. Tobouti PL, et al. Benign vascular lesions of the lips: Diagnostic approach: J Cutan Pathol; 2017;44(5):451-5. PubMed
4. Calonje E, MacKee PH, editores. McKee’s pathology of the skin: with clinical correlations ; 4. ed. Edinburgh: Elsevier; 2012.
CASO 88
15 de noviembre
Dra. Manuela Munera. Residente de Dermatología. UPB. Medellín , Colombia
Dra. Camila Montoya. Dermatopatóloga. Asunción. Paraguay
Dr. Rodrigo Restrepo. Profesor Programa de Dermatopatología. Universidad CES. Medellín , Colombia
Hombre, 28 años. Lesión tumoral en rodilla derecha. 8 meses de evolución.
Respuestas de los participantes comienzan a aparecer el 20 Noviembre . Solución y cierre de caso 30 de noviembre 2018
 F1Panorámica, formación exófitica en domo, presencia de proliferación melanocitica dermoepidérmica, con asimetría |  F2Panóramica, mayor detalle. |  Detalle de la proliferación melanocítica, dermo-epidérmica. Epidermis con nidos de células fusiformes y disposición irregular. Dermis con nidos fusiformes e epitelioides. |
|---|---|---|
 F3Mayor detalle del componente epidérmico. Izquierda. Nidos de células grandes epitelioides. Derecha: Nidos de melanocitos en la unión, constituidos por células grandes, con núcleos vesiculosos y nucléolos prominentes. |  F4Composición de fotos: Mitosis en el componente epidérmico (arriba izquierda). Mitosis en el componente dérmico ( izquierda abajo; derecha arriba y abajo) |  F5Composición de fotos: Mitosis Atípicas en el componente dermis (izquierda) . Mayor detalle (derecha). |
 F7SOX-10. Positivo, nuclear fuerte difuso en melanocitos epidérmicos y dérmicos. |  F8Ki-67. indice de proliferación elevado, positividad difusa, ausencia de gradiente. |  F9P21. Positividad variable en los melanocitos del componente dérmico. |
 F10P16. Negativo en los melanocitos del componente epidérmico y dérmico. |
Con las características histológicas observadas, el diagnóstico más probable es:
A. Reticulohistiocitoma solitario
B. Lesión Spitzoide atípica
C. Melanoma Spitzoide
D. Nevus de Spitz
Respuestas
Melanoma Spitzoide Beatriz di Martino
Melanoma Spitzoide Magda Jiménez
Melanoma Spitzoide Andrés Fabían López R.
Melanoma Spitzoide Luis Fdo. Cárdenas C.
Les. Spitzoide Atípica Sandro Casavilca
Melanoma Spitzoide Juan M. Gonzáles
Melanoma Spitzoide Griselda de Anda
Les. Spitzoide Atípica María del Valle Centeno
Les. Spitzoide Atípica Camila Otero
Melanoma Spitzoide Fernando Cabo Gómez
Melanoma Spitzoide Luis H. Cruz Contreras
Melanoma Spitzoide Esther Mariela Estrada
Melanoma Spitzoide Elizabeth Ball de P.
Melanoma Spitzoide Pascual Meseguer Garcia
Melanoma Spitzoide José Manuel Rodríguez G.
Melanoma Spitzoide Guillermo Ramos
Melanoma Spitzoide Gloria Mendoza
Melanoma Spitzoide Clara Jaramillo
Melanoma Spitzoide Carlos Cortés
Respuesta
C.Melanoma Spitzoide (correcta):
Melanoma que tiene similitud tanto clínica como histológica con el nevus de Spitz. Clínicamente se presentan como pápulas o nódulos que pueden ser o no pigmentados. Histológicamente, muestran citomorfología fusiforme o epitelioide asociados a hiperplasia epidérmica (F1-4). La presencia de asimetría (F1-2), alteración en la maduración, mitosis profundas tipicas(F5) y atípicas (F6), necrosis, diseminación pagetoide y/o pleomorfismo celular apoyan el diagnóstico de Melanoma. En cuánto a la inmunohistoquímica el Ki-67 es un marcador importante para diferenciarlo del nevus de Spitz y de lesiones Spitzoides, siendo mayor al 10% y difuso, en el melanoma; el HMB45 no muestra gradiente (F8); el P21 es bajo (F9) y el P16 usualmente es negativo (F10).
El pronóstico es comparable con otras variantes de melanoma de igual Breslow y este varía según la edad del paciente siendo mejor en niños y adolescentes.
A. Reticulohistiocitoma solitario (incorrecta): también conocido como histiocitoma epitelioide solitario, es un tumor dérmico benigno. Clínicamente pueden ser nódulos únicos o múltiples, en adultos jóvenes. Histológicamente presenta histiocitos grandes con citoplasma eosinofílico con apariencia en vidrio esmerilado en la dermis superior y media, con infiltrado inflamatorio mixto y puede presentar leve atipia nuclear y actividad mitótica baja. En la inmunohistoquimica es positivo para CD68, CD163 y vimentina; variable para MITF, S100; y negativo para CD1a, citoqueratina, HMB 45 y Melan-A.
B. Lesión Spitzoide atípica (incorrecta): estas lesiones se encuentran en el espectro entre los nevus de Spitz y melanoma Spitzoide.
D. Nevus de Spitz (incorrecta): histológicamente es una lesión simétrica, bien delimitada en forma de domo, epidermis con acantosis y retracción entre los nidos de melanocitos y la epidermis. Pueden observarse cuerpos de kamino, que no son patognomónicos. El P16 es positivo y homogéneo, el HMB-45 muestra gradiente y el índice de proliferación es bajo, Ki-67 (0-2%).
Bibliografía:
1. Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 17 de marzo de 2014;20(3). PubMed
2. Stefanaki C, Stefanaki K, Chardalias L, Soura E, Stratigos A. Differential diagnosis of Spitzoid melanocytic neoplasms. J Eur Acad Dermatol Venereol. agosto de 2016;30(8):1269-77 PubMed
CASO 87
31 de octubre
Dra. Paula Abelenda. Dermatóloga. Montevideo , Uruguay
Dra. Camila Montoya Dermatopatóloga. Asunción. Paraguay
Dr. Rodrigo Restrepo. Profesor del Programa de Dermatopatología. Universidad CES. Medellín , Colombia
Mujer, 35 años. Placas alopécicas pruginosas en cuero cabelludo, 5 años de evolución. Pull test (+) en anágeno.
Respuestas de los participantes comienzan a aparecer el 10 Noviembre . Solución y cierre de caso 15 de noviembre 2018
 Ausencia de orificios foliculares. Eritema y descamación perifolicular. |  |  |
|---|---|---|
 |  |  |
 |  |  |
Respuestas
Liquen plano pilar Beatriz di Martino
Foliculitis decalvans Márcio Martins Lobo J.
Liquen plano pilar Víctor Manuel Delgado O.
Lupus discoide Juan
Liquen plano pilar Helga Sarti
Liquen plano pilar Anónimo
Foliculitis decalvans Fernando Cabo Gómez
Liquen plano pilar Pascual Meseguer García
Liquen plano pilar Guillermo Ramos R.
Liquen plano pilar Esther Mariela E.
Liquen plano pilar Regina Barros D.
Liquen plano pilar María Gloria Mendoza
Liquen plano pilar María Catalina Cuéllar
Lupus discoide Griselda de Anda
Liquen plano pilar Juan M. Gonzáles
Liquen plano pilar Eduwiges Martínez L.
Liquen plano pilar Juan C. Bonilla Jassir
Liquen plano pilar Diego Díaz
Con las características clínicas e histológicas observadas el diagnóstico más probable es:
A. Lupus discoide
B. Liquen plano pilar
C. Lues
D. Foliculitis decalvans
Respuesta
Caso con elementos clínicos e histopatológicos superpuestos. En definitiva, es una alopecia cicatricial tanto clínica (F1) como histológicamente. Con pérdida de glándulas sebáceas, fibrosis, predominio de infiltrado linfocítico (F2, F4) y descamación prematura de la vaina radicular interna (F6) (1).
B. Liquen plano pilar (Correcto): La fibrosis y el infiltrado inflamatorio están localizados alrededor del segmento superior sin afectar la epidermis (F1-F4). La dermis interfolicular está en general indemne (F1, F2), a no ser que haya dos folículos adyacentes (F4). Hay tendencia a fusionar hasta 3 estructuras foliculares, lo cual no es muy usual en este grupo de alopecias linfocíticas. Sin embargo, este fenómeno puede observarse en casos crónicos (F5). La fibroplasia lamelar y daño de la interfase entre el folículo y la dermis, con apoptosis residual es aparente (F6, F7) (2).
A.Lupus discoide (LD) (Incorrecto): Es difícil que el LD no comprometa la epidermis ni los anexos y en general su afectación de la dermis es difusa, superficial y profunda, a veces con mucina visible.
C. Lues: La lues no produce los cambios descritos de alopecia cicatricial enumerados al principio, pero llama la atención la presencia de plasmocitos en las estelas foliculares profundas (F8, F9). El VDRL fue negativo y el cuadro clínico no corresponde a una alopecia sifilítica en sus formas reconocidas (sintomática y esencial). La presencia de plasmocitos es intrigante, aunque ha sido descrito cada vez más en alopecias de diferentes tipos (lupus, alopecias cicatriciales con ruptura folicular, psoriasis asociada al uso de anti- factor de necrosis tumoral alfa, etc.).
D. Foliculitis decalvans: No hay infiltrado de neutrófilos en la superficie y el fenómeno de pelo de muñeca no es muy aparente clínicamente.
Bibliografia
1. Olsen EA, Bergfeld WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)-Sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol 2003;48: 103–10. PubMed
2. Soares VC, Mulinari-Brenner F, Souza TE. Lichen planopilaris epidemiology: a retrospective study of 80 cases. An Bras Dermatol. 2015 Sep-Oct;90(5):666-70. Free text link
3. Bolduc, C., Sperling, L. C., & Shapiro, J. (2016). Primary cicatricial alopecia. Journal of the American Academy of Dermatology, 75(6), 1081–1099.
CASO 86
15 de octubre
Dra. Liza Arias. Residente de Dermatopatología. Programa de Dermatopatología. Universidad CES. Medellín, Colombia.
Dr. Camila Montoya. Dermatopatóloga. Universidad CES. Medellín.
Mujer de 69 años, lesión pigmentada en muslo.
Respuestas de los participantes comienzan a aparecer el 20 octubre. Solución y cierre de caso 31 de octubre 2018
 F2Placa marrón-negro de 2.5 cms, asimétrica y de bordes irregulares |  F2Red de pigmento atípica, múltiples colores, estructuras blanco-azul, proyecciones radiales y múltiples puntos marrones de manera focal |  F3Panorámica. Lesión melanocítica asimétrica que involucra epidermis sin invadir dermis |
|---|---|---|
 F4Mayor detalle. Melanocitos atípicos solitarios y en nidos con diseminación pagetoide a estratos superiores de la epidermis |  F5Melanocitos atípicos solitarios y en nidos con diseminación pagetoide a estratos superiores de la epidermis | 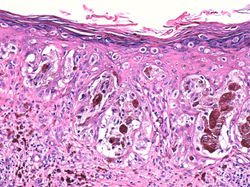 F6Melanocitos atípicos solitarios y en nidos con diseminación pagetoide a estratos superiores de la epidermis |
 F7Mayor aumento. Melanocitos atípicos solitarios y en nidos con diseminación pagetoide a estratos superiores de la epidermis |  F8MITF-1 positivo en los núcleos de los melanocitos atípics con ee diseminación pagetoide |  F9MITF-1, mayor detalle positividad nuclear en los melanocitos atípicos. |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Nevus melanocítico congénito
B. Nevus de Spitz
C. Enfermedad de Paget extra mamaria pigmentada
D. Melanoma In situ
Respuestas
Melanoma in situ Maria Antonieta Tirado
Melanoma in situ Melanie Marmolejo Chavira
Melanoma in situ Carolina Delgado
Melanoma in situ Andrea Castaño
Melanoma in situ Fernando Cabo Gómez
Melanoma in situ Félix Padilla
Melanoma in situ Marly Saads
Enfermd. de Paget Alvaro Ibarra
Melanoma in situ Julia Inés Mesa
Nevus de Spitz Magda Jimena Vargas Díaz
Melanoma in situ Guillermo Ramos Rodríguez
Melanoma in situ Griselda de Anda
Melanoma in situ Carolina Cabrera Salom
Melanoma in situ Allison Zarbo de Gómez
Melanoma in situ Carlos Cortés Caballerro
Melanoma in situ Carlos Ronald Martínez C
Melanoma in situ María del Consuelo Gómez
Melanoma in situ Nohemy Correa López
Enfermd. de Paget Montse Molgo
Respuesta
D. Melanoma in situ (correcta)
Mácula o placa que suele medir más de 6mm, con asimetría y más de un color (F1). Dermatoscópicamente muestra red de pigmento atípica, múltiples colores, patrón gris-azul, entre otros. (F2)(1) Histológicamente es una proliferación de melanocitos atípicos en la epidermis sin invasión a la dermis siendo una lesión mal circunscrita, con predominio de melanocitos solitarios, borramiento de las crestas epidérmicas, diseminación pagetoide y nidos hacia la capa granulosa y estrato córneo, con consumo de la epidermis y compromiso anexial (F3-F7). MITF-1 marca el núcleo de los melanocitos (F8-F9)(2).
A. Nevus melanocítico congénito (incorrecta): Los hallazgos dependen de la edad del paciente. En neonatos puede observarse diseminación pagetoide así como atipia citológica siendo frecuente aún en la periferia de la lesión. El Componente dérmico presenta melanocitos con adecuada maduración. Los anexos se ven rodeados por células névicas. (3)
B. Nevus de Spitz (incorrecta): Histológicamente se observa una lesión circunscrita y simétrica, células névicas epitelioides y/o fusiformes. Pueden presentar diseminación pagetoide en la porción central, mitosis cerca de la capa basal epidérmica hasta en el tercio superior de la dermis, con áreas de maduración irregular. Es uno de los principales diagnósticos diferenciales de melanoma. (4). Ver caso 66
C. Enfermedad de Paget extramamaria pigmentada (incorrecta): Clínica e histológicamente puede ser indistinguible del melanoma por lo que precisa inmunohistoquímica para su diferenciación. Se observan células de citoplasma claro, núcleos vesiculosos y nucléolos prominentes con migración transepidérmica(5). Ver caso 61
Revisión bibliográfica
-
Higgins W., Lee K., Galan A., et al. Melanoma in situ. J Am Acad Dermatol. 2015;73(2):181-190. PubMed
-
Higgins W., Lee K., Galan A., et al. Melanoma in situ. J Am Acad Dermatol. 2015;73(2):193-203. PubMed
-
Olivera A. Riesgo de melanomas sobren nevos melanocíticos congénitos. Arch. Argent. Dermatol. 2012;62:211-218. FreeTextLink
-
Verardino GC., Rochael MC. Spitz nevi in the classic histopathological pattern-lamb in wolf´s clothing. An Bras Dermatol. 2015;90(1):91-5. PubMed
-
Monti F., Caruso A., Garay I. Enfermedad de Paget extramamaria axilar: presentación de un caso y revisión de literatura. Med Cutan Iber Lat Am. 2017;45(1):25-28. PubMed
CASO 85
1 de octubre
Dra. Mónica Salazar. Residente de Dermatopatología. Programa de Dermatopatología. Universidad CES. Medellín, Colombia.
Dra. Camila Montoya. Dermatopatológa. Universidad CES. Medellín.
Mujer, 20 años. Lesión nodular marrón rojiza en hombro, 1.0 cm de diámetro. Asintomática.
Respuestas de los participantes comienzan a aparecer el 10 octubre. Solución y cierre de caso 15 de octubre 2018
 F1Lesión formada por un retículo pigmentado delicado en periferia y un parche blanco central, en huevo frito, asociado a un patrón vascular polimorfo. |  F2Imagen panorámica, lesión dérmica fusocelular bien delimitada |  F3Mayor detalle, haces de colágeno engrosados, hilinizados |
|---|---|---|
 F4Detalle de haces de colágeno hialinizados |  F5Detalle de los haces de colágeno engrosados disposición irregular (izquierda y centro), algunos adoptan patrón circular (derecha) |  F6Haces de colágeno rodeados por células fusiformes |
 F7Haces de colágeno rodeados por células fusiformes |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Cicatriz hipertrófica
B. Dermatofibroma queloidiano
C. Fasceitis nodular queloidiana
D. Queloide
Respuestas
Dermatofibroma queloidiano Marcelo Toro
Dermatofibroma queloidiano Verónica Posso
Dermatofibroma queloidiano Sofía Rolldán
Dermatofibroma queloidiano Regina Barros D.
Dermatofibroma queloidiano Angela Seidel
Dermatofibroma queloidiano Beatriz di Martino
Dermatofibroma queloidiano Francisco Chavez R.
Dermatofibroma queloidiano Anónimo
Dermatofibroma queloidiano Sandro Casavilca Z.
Fasceitis nodular queloidiana Marcela Gómez
Dermatofibroma queloidiano Lina M. Velásquez T.
Dermatofibroma queloidiano Julieta Corral
Dermatofibroma queloidiano Alvaro Peynado
Dermatofibroma queloidiano Luis H. Cruz Contreras
Dermatofibroma queloidiano Julia Mesa
Dermatofibroma queloidiano Ronald Martinez
Dermatofibroma queloidiano Andres Castaño V.
Dermatofibroma queloidiano María Janeth Vargas
Dermatofibroma queloidiano Carolina Cabrera S.
Dermatofibroma queloidiano M. del Consuelo Gómez
Dermatofibroma queloidiano Guillermo Ramos R.
Dermatofibroma queloidiano Guiselle Romero Caimi
Dermatofibroma queloidiano Carlos Cortés C.
Respuesta:
B. Dermatofibroma queloidiano
Es un tumor fibrohistiocítico benigno, más frecuente en mujeres jóvenes, con predilección por extremidades inferiores. Clínicamente pueden ser lesiones únicas o múltiples de aspecto papular, en placa o nodular e incluso pediculadas. El color es variable desde el eritemato-parduzco hasta violáceo, rojizo o amarillento. La mayoría presenta el "signo del hoyuelo" que consiste en el deslizamiento de la lesión hacia planos profundos al ejercer presión lateralmente, dando la impresión de que el tumor se “escapa” (F1)(1,3).
Histológicamente en la forma clásica se presenta como lesiones dérmicas bien definidas, compuestas por células fusiformes entrelazadas, distribuidas al azar algunas veces con patrón estoriforme, asociados a presencia de hiperplasia de la epidermis suprayacente e hiperpigmentación de la capa basal, este fenómeno conocido como inducción epidérmica es característico de este tipo de lesiones. La variante queloidiana del dermatofibroma, clínicamente indistinguible de la forma clásica, presenta la típica proliferación fusocelular irregular asociada a áreas circunscritas compuestas por haces de colágeno gruesos intensamente eosinofílicos generalmente en la porción superficial con una orientación irregular (F2-7) observarse también haces de colágeno dispuestos de forma circular (F5) (2).
A. Cicatriz hipertrófica (Incorrecto): Las fibras de colágeno hialinizadas son menos prominentes y a veces se acompaña de una reacción granulomatosa de cuerpo extraño.
C. Fascitis nodular (Incorrecto): Lesión relativamente bien circunscrita, no encapsulada, compuesta por fibroblastos fusiformes o redondeados que se disponen aleatoriamente o agrupados en forma de S. Las mitosis son frecuentes pero no atípicas. Se han descrito varios subtipos histológicos basados en la celularidad, el estroma mixoide y la presencia de colágeno.
D. Queloide (Incorrecto): Proliferación fibroblástica nodular con haces de colágeno hialinizadas hipocelulares, eosinófilicas en la dermis superficial y profunda.
Bibliografía:
-
Bandyopadhyay MR, Besra M, Dutta S, et al. Dermatofibroma: Atypical Presentations. Indian J Dermatol. 2016; 61:121. Free Full Text
-
Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol. 1998;22:564–8 PubMed
-
Kim J, Cho H, Moon S. Rare experience of keloidal dermatofibroma of forehead. Arch Craniofac Surg. 2018; 19:72-74. PubMed
CASO 84
15 de Septiembre
Dra. Liza arias. Residente de Dermatopatología. Dr. Andrés Flórez. Jefe Programa Dermatopatología. Dra. Camila Montoya. Dermatopatológa. Programa de Dermatopatología. Universidad CES. Medellín, Colombia.
Mujer con lesiones ampollosas en ambos dorsos de manos y antebrazo.
Respuestas de los participantes comienzan a aparecer el 20 de Septiembre. Solución y cierre de caso 30 de septiembre 2018
 F1Presencia de vesículas, eritema y costras en dorso de las manos |  F2Panorámica, Ampolla subepidérmica |  F3Ampolla subepidérmica con leve infiltrado inflamatorio |
|---|---|---|
 F4Ampolla subepidérmica y festonamiento de las papilas dérmicas |  F5Festonamiento de las papilas dérmicas |  F6Festonamiento de las papilas dérmicas |
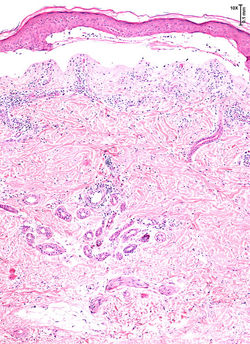 F7Ampolla subepidérmica, festonamiento de las papilas e infiltrado inflamatorio |  F8Infiltrado inflamatorio compuesto por linfocitos y eosinófilos |  F9Presencia de material eosinofílico en la epidermis |
 F10Engrosamiento de las paredes vasculares |  F11Engrosamiento de las paredes vasculares |  F12 PAS-DTinción de PAS que resalta material hialino alrededor de los vasos y en la unión dermoepidérmica |
 F13 PAS-DTinción de PAS que resalta material hialino alrededor de los vasos y en la unión dermoepidérmica |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Epidermolisis ampollosa adquirida
B. Penfigoide ampolloso
C. Amiloidosis ampollosa
D. Porfiria
A los pocos días de haber colocado este caso experimentamos algunas dificultades para enviar y recibir su opinión.
Afortunadamente fueron solucionados y ya usted puede darnos su diagnóstico sin ningún problema.
Respuestas
Porfiria María Janeth Vargas
Porfiria Julia Mesa
Porfiria Daniela Gómez Osorio
Porfiria Diego Díaz
Porfiria Márcio Martnis Lobo Jardim
Porfiria Carolina Cabrera Salomón
Porfiria Gerardo Prada Ch.
Porfiria María del Consuelo Gómez
Porfiria Guillermo Ramos R,
Porfiria María del Valle Centeno
Porfiria Beatrz di Martino
Porfiria Bárbara Sáenz
Respuesta
D. Porfiria cutánea tarda (correcta):
Es un desorden metabólico de la biosíntesis del grupo hemo causado por una deficiencia catalítica en la descarboxilasa uroporfirinógeno. Existen múltiples disparadores ambientales y genéticos por lo que pueden observarse formas adquiridas, heredadas y tóxicas. Las manifestaciones cutáneas se producen en áreas fotoexpuestas como el dorso de las manos tal como observamos en nuestro paciente (F1) e incluyen fragilidad, formación de vesículas-ampollas, cicatrices atróficas, hiperpigmentación post-inflamatoria, hipertricosis y milia, entre otras. Histopatológicamente se observa formación de ampolla subepidérmica (F2-4), festonamiento de las papilas dérmicas (F5-6) y un infiltrado inflamatorio leve o ausente (F7-11). La tinción de PAS resalta el material hialino en las paredes de los vasos dérmicos superficiales y en la unión dermoepidérmica que se deposita como respuesta a episodios repetidos de daño a las pared de los vasos (F12-13).
A. Epidermolisis ampollosa adquirida (incorrecta): enfermedad autoinmune mecano-bulosa que afecta la unión dermo-epidérmica de las superficies acrales sujetas a trauma constante. Se observan vesículas, ampollas y milia. La histología muestra ampolla subepidérmica, y según la variante; puede observarse desde ausencia de infiltrado hasta infiltrado inflamatorio compuesto por neutrófilos, eosinófilos, monocitos y linfocitos. Puede haber presencia de fibrosis (2,3).
B. Amiloidosis ampollosa (incorrecta): variante rara del liquen amiloide y en su mayoría se asocia a involucro sistémico. La histología demuestra ampolla subepidérmica y depósitos hialinos de amiloide en la base de la ampolla(4).
C. Penfigoide ampolloso (incorrecta): enfermedad ampollosa autoinmune más común que afecta a personas de edad avanzada en su mayoría. Histológicamente hay presencia de ampolla subepidérmica con un grado variable de infiltrado inflamatorio compuesto por linfocitos, neutrófilos y característicamente por eosinófilos(5). Ver Caso 9
Bibliografía
-
Vieira FMJ., Martin JEC. Porphyria Cutanea Tarda. An Bras Dermatol. 2006;81(6):569-80. FreeTextLink
-
Cyr J., Liu A., Ghazarian D., et al. Case Report of a 21-year-old man with epidermolysis bullosa acquisita. J Cutan Med Surg. 2018;22(3):356-358 PubMed
-
Kridin K. Accesible diagnostic methods to differentiate between epidermolysis bullosa acquisita and other subepidermal autoimmune bullous disease. Indian J Dermatol. 2018;63(5):445-48 PubMed FreeText
-
Suranagi VV., Siddramappa B., Bannur HH., et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60(1):105 PubMed Freetext
-
Amber K., Valdebran M., Kridin K., et al. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med. 2018;5:1-15 PubMed FreeText
CASO 83
1 de Septiembre
Dra. Liza arias y Camila Montoya Bueno. Residente de Dermatopatología. Programa Dermatopatología.
Dr. Rodrigo Restrepo. Profesor Programa Dermatopatología. Universidad CES. Medellín, Colombia.
Joven con lesiones hiperqueratósicas lineales que se concentran en axilas y pliegues. No hay historia familiar.
Respuestas de los participantes comienzan a aparecer el 15 de Septiembre. Solución y cierre de caso 15 de Agosto 2018
 F1Pápulas y placas verrucosas que confluyen siguiendo las líneas de Blashcko de manera bilateral |  F2Pápulas y placas verrucosas que confluyen siguiendo las líneas de Blashcko |  Panóramica, acantosis con hiperqueratosis compacta |
|---|---|---|
 F4Marcada hiperqueratosis compacta |  F5Vacuolización perinuclear de los queratinocitos del estrato espinoso y granuloso con aumento del tamaño de los gránulos de queratohialina |  F6Vacuolización perinuclear de los queratinocitos del estrato espinoso y granuloso con aumento del tamaño de los gránulos de queratohialina |
 F7mayor detalle, vacuolización perinuclear de los queratinocitos del estrato espinoso y granuloso |  F8Mayor detalle del estrato espinoso y granuloso con aumento del tamaño de los gránulos de queratohialina |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Ictiosis vulgar
B. Penfigo foliáceo
C. Nevus epidermolítico
D. Enfermedad de Darier
Respuestas
Nevus epidermolítico María Janeth Vargas
Nevus epidermolítico Beatriz di Martino
Enfermedad de Darier Anónimo
Nevus epidermolítico Marcio Martins Lobo J.
Enfermedad de Darier Roniel Contreras
Nevus epidermolítico Marcela Olaya
Nevus epidermolítico Francisco Chávez Rojas
Enfermedad de Darier Juan F. Pinilla
Nevus epidermolítico Mónica Zapata
Nevus epidermolítico Juan M. González
Nevus epidermolítico Regina Barros Domínguez
Nevus epidermolítico Esther Mariera Estrada
Nevus epidermolítico Luis Humberto Cruz
Ictiosis vulgar Sandra Quijano Moreno
Nevus epidermolítico Mario Lima
Nevus epidermolítico Guillermo Ramos R.
Enfermedad de Darier Marly Saads Fonseca
Nevus epidermolítico Carmen López Acosta
Nevus epidermolítico María del Valle Centeno
Nevus epidermolítico Julia Mesa
Nevus epidermolítico Alvaro Peynado
Nevus epidermolítico Jennifer Castillo G.
Respuesta
C. Nevus epidermolítico (correcta)
Hamartoma cutáneo inusual que resulta de una mutación post-cigótica afectando las citoqueratinas 1 y 10. Esto puede traducirse como un mosaicismo de la hiperqueratosis epidermolítica por lo que se conoce también como eritrodermia ictiosiforme nevoide bulosa. (1) Clínicamente se presenta como pápulas y placas verrucosas de color marrón que confluyen y aparecen en las extremidades a lo largo de las líneas de Blashcko. Puede ser unilateral o presentarse de forma bilateral como nuestro caso. (F1-2)
Histológicamente se observa hiperqueratosis compacta (F3-4), vacuolización perinuclear de los queratinocitos del estrato espinoso y granuloso, aumento del tamaño de los gránulos de queratohialina y un infiltrado inflamatorio leve mononuclear perivascular. (F5-8) (2)
A. Ictiosis vulgar (incorrecta): desorden genético cutáneo descamativo más frecuente del grupo de las ictiosis. Se presenta como descamación fina que va de leve a moderada, respetando las áreas flexurales y puede encontrarse junto con la dermatitis atópica.
Histológicamente se observa hiper-ortoqueratosis, epidermis hiperplásica, normal o atrófica y un estrato granuloso delgado o ausente. (3)
B. Pénfigo foliáceo (incorrecta): enfermedad autoinmune ampollosa que principalmente afecta las regiones seborreicas .
Histológicamente se observa clivaje subcórneo (dentro o debajo de la capa granular) con presencia de queratinocitos acantolíticos. (4)
D. Enfermedad de Darier (incorrecta): desorden autosómico dominante que compromete la adhesión intercelular. Clínicamente se presenta como pápulas hiperqueratósicas y oleosas en una distribución seborreica. Histológicamente se observa acantolisis suprabasal y disqueratosis con presencia de cuerpos redondos y granos.
Bibligrafía:
-
Mishra V.. et al. Bilateral Systematized Epidermolytic Verrucous Nevus: A rare entity. Indian J Dermatol 2015;60(4):397-399 Pubmed
-
Scherli F, et al. Nevo epidermolítico: ictiosis queratinopática menor. Med Cutan Iber Lat Am 2016;44(1):45-47 Pubmed
-
Krug M, et al. Ichthyosis Part 1: Differential diagnosis of vulgar ichthyosis and therapeutic options. J Dtsch Dermatol Ges 2009;7(6):511-19 Pubmed
-
Hans-Filho G., Aoki V., Bittner N., et al. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol 2018;93(5):638-50 Pubmed
-
Pettit C., Ulman C., Spohn G., et al. A case of segmental Darier disease treated with doxycycline monotherapy. Dermatol Online J 2018;(24)3:1-3 Pubmed
CASO 82
15 de Agosto
Dra. Mónica Salazar y Camila Montoya Bueno. Residente Programa Dermatopatología.
Dr. Andres Flórez. Coordinador Programa Dermatopatología. Universidad CES. Medellín, Colombia.
Sexo femenino, 2 años. Madre con psoriasis. Presenta desde hace 20 días pápulas parduzcas hiperqueratósicas en ambos pliegues inguinales, asintomáticas.
Respuestas de los participantes comienzan a aparecer el 25 de Agosto. Solución y cierre de caso 31 de Agosto 2018
 |  |  |
|---|---|---|
 |  |  |
 |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Dermatitis de contacto
B. Paraqueratosis granular
C. Psoriasis invertida
D. Dermatofitosis
Respuestas
Dermatofitosis Anónimo
Paraqueratosis granular María Janeth Vargas
Paraqueratosis granular María Eugenia Seminario
Paraqueratosis granular Susan Peralta Montes
Paraqueratosis granular Diego Díaz
Paraqueratosis granular Márcio Martins Lobo Jardim
Dermatofitosis Laura Carreño Toro
Paraqueratosis granular Jenniffer Castillo Guarnizo
Paraqueratosis granular Malena Ortiz Galarza
Paraqueratosis granular Daniela Gómez Osorio
Psoriasis invertida Ismery Cabello
Paraqueratosis granular Regina Barros Domingues
Paraqueratosis granular Carmen López Acosta
Paraqueratosis granular Guillermo Ramos
Paraqueratosis granular Francisco Chávez Rojas
Psoriasis invertida María del Consuelo Gómez
Paraqueratosis granular Melanie Marmolejo Chavira
Paraqueratosis granular Carlos Cortés C.
Dermatofitosis Marcela Vasconcelos
Respuesta
B. Paraqueratosis granular (correcta):
Desorden adquirido de la queratinización, descrito por primera vez en 1991 por Northcutt con el nombre de "paraqueratosis granular axilar" (1). Su frecuencia es rara y afecta predominantemente a mujeres de mediana edad. Sin embargo, se han descrito casos en infantes. Se desconoce su etiología pero se ha relacionado con reacciones de contacto con antitranspirantes, oclusión, uso excesivo de cremas y obesidad. En lactantes el uso de pañales parece jugar un papel importante en su aparición. (2,4).
Clínicamente presenta pápulas y placas hiperqueratósicas, eritemato-parduzcas asintomáticas, ocasionalmente pruriginosas. Generalmente se localiza en axilas (F1-2) pero se ha reportado también en pliegues submamarios, ingles, vulva y región perianal, así como en áreas no intertriginosas como abdomen y región lumbosacra. En casos infantiles se han descrito dos patrones clínicos: placas lineales bilaterales en los pliegues inguinales y placas geométricas eritematosas que subyacen a los puntos de presión del pañal(2).
La histología es característica de ahí el nombre de esta entidad, observándose un engrosamiento de la capa córnea con paraqueratosis compacta y persistencia de gránulos de queratohialina, con estrato granuloso preservado(3) (F3-8). Se ha informado de paraqueratosis granular incidental asociada con molusco contagioso, dermatomiositis, infección por dermatofitos y carcinomas(4). Nuevas técnicas de diagnóstico han sido propuestas, como el examen citológico de los raspados superficiales de las lesiones. (3).
A. Dermatitis de contacto (Incorrecto): A la histología muestra una dermatitis espongiótica (aguda, subaguda o crónica)
C. Psoriasis Invertida (Incorrecto): Los hallazgos son variables dependiendo de la evolución de la lesión, presentando una dermatitis psoriasiforme regular con paraqueratosis, disminución o ausencia de la capa granular, dilatación de los vasos capilares en la dermis papilar, microabscesos de Munro y pústulas de Kogoj.
D. Dermatofitosis (incorrecto): Se observa hifas y levaduras en el estrato córneo, ademas de paraqueratosis y colecciones de neutrófilos.
BIibliografia:
-
Braun M, Laaff H. Granular parakeratosis-a clinical-pathological correlation of 10 cases. J Dtsch Dermatol Ges. 2009 Apr;7(4):340-4. PubMed
-
Chang M, Kaufmann J, Orlow S, et al. Infantile granular parakeratosis: recognition of two clinical patterns. J Am Acad Dermatol. 2004 May;50(5 Suppl):S93-6. PubMed
-
Deniz A, Yasemin O, Ozlem A. Infantile Granular Parakeratosis: Cytologic Examination of Superficial Scrapings as an Aid to Diagnosis. Pediatric Dermatology.2015; 32(3): 392–396. PubMed
-
Yang J, Lee H, Noh T, et al. Granular parakeratosis of eccrine ostia. Ann Dermatol. 2012 May;24(2):203-5. PubMed
CASO 81
1 de Agosto
Dra. Liza Arias y Camila Montoya. Residentes de Dermatopatología. Programa Dermatopatología
Hombre, 48 años. Múltiples pápulas en piernas, intensamente pruriginosas.
Respuestas de los participantes comienzan a aparecer el 6 de Agosto. Solución y cierre de caso 15 de Agosto 2018
 F1Múltiples pápulas pigmentadas formando placas en miembros inferiores. Cortesía Dr. E. Peña, Dermatólogo. Cl. Mdlln. Col. |  F2Pápulas confluentes, algunas con costra hemática secundaria a rascado. |  F3Pápulas queratósicas y confluentes. |
|---|---|---|
 F4Epidermis con hiperqueratosis y elongación de las crestas |  F5Papilas dérmicas ensanchadas | 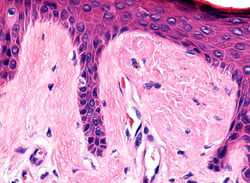 F5Incontinencia de pigmento y material globular eosinofílico |
 F6Incontinencia de pigmento y material globular eosinofílico |  F(Material globular consistente con amiloide localizado en las papilas resaltado con cristal violeta y rojo congo. |  F9 Rojo congo - DetalleMaterial globular consistente con amiloide localizado en las papilas resaltado con rojo congo. |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Liquen amiloide
B. Mucinosis papuar
C. Liquen plano hipetrófico
D. Amiloidosis macular
Respuestas
Liquen amiloide Francisco Chávez R
Liquen amiloide María Janeth Vargas
Liquen amiloide Griselda de Anda
Liquen amiloide Sandra Quijano Moreno
Liquen amiloide Marly Saads Fonseca
Liquen amiloide Beatriz di Martino
Liquen amiloide Julieta Corral
Liquen amiloide Dante Villamil
Liquen amiloide Cibele Conceição dos Apóstolos
Amiloidosis macular Juan M. González
Liquen amiloide Eduardo Gómez T.
Amiloidosis macular Cristielle
Liquen amiloide Sofía Roldán
Liquen amiloide Diego Díaz
Liquen amiloide Susan Peralta Montes
Amiloidosis macular Malena Ortiz Galarza
Liquen amiloide Juan Felipe Pinilla
Mucinosis papular Jorge Ospina
Liquen amiloide Gerardo Prada
Liquen amiloide Giselle Romero Caimi
Liquen amiloide Jenniffer Castillo G.
Liquen amiloide Paola M. L.Chávez T.
Liquen amiloide Naty Lazcano
Amiloidosis macular Carlos Cortés Caballero
Liquen amiloide Pilar Alemán
Liquen amiloide Kenneth Salazar
Liquen amiloide Maribel Cortez
Liquen amiloide Maria Eugenia Seminario
Respuesta
A. Liquen amiloide (correcta): Es una forma de amiloidosis cutánea primaria localizada. Se presenta con múltiples pápulas firmes, hiperqueratósicas y pigmentadas que con el tiempo forman placas gruesas resistentes al tratamiento (F1-3). Clinicamente cursa con intenso prurito y su prinicipal localización son las extremidades inferiores. Usualmente es hallazgo único, pero puede asociarse a MEN2A(1). La histopatología muestra epidermis con hiperqueratosis, vacuolización de las células basales, acantosis y elongación de las crestas (F4). La dermis presenta depósitos globulares eosinofílicos de amiloide, incontinencia de pigmento e infiltrado inflamatorio linfocitario perivascular superficial (2) (F5-7). Este material produce reacción metacromática con cristal violeta (3) y es positiva para rojo congo con una sensibilidad de 89% (2) (F8-9).
B. Mucinosis papular (incorrecta): Se observan pápulas pequeñas localizadas en miembros superiores. Histológicamente muestran acúmulos de mucina en la dermis reticular superior con separación de las fibras de colágeno (4).
C. Liquen plano hipertrófico (incorrecta): Son placas hiperqueratósicas gruesas con escama fina adherente, pruriginosas, usualmente localizadas en ambos miembros inferiores. Histológicamente se observa hiperplasia epidérmica, acantosis, hipergranulosis e hiperqueratosis lamelar compacta centrándose en el infundíbulo folicular y acrosiringio. El daño basal está confinado a las puntas de las crestas y el infiltrado en banda se encuentra ausente (5).
D. Amiloidosis macular (incorrecta): Se presenta como máculas hiperpigmentadas que pueden ser confluentes. Pueden ser o no sintomáticas y son mas frecuentes en las áreas extensoras de los brazos y espalda superior. El depósito de amiloide tiende a ser escaso observándose como glóbulos eosinofílicos pequeños (3).
Bibliografia:
1. Ladizinski B., Lee K. Lichen Amyloidosis. CMAJ. 2014;187(7):532 Pubmed
2. Mehrotra K.,. et al. Primary Cutaneous Amyloidosis: a clinical, histopathological and immunofluorescence study. Journal of Clinical and Diagnostic Research. 2017:11(8);WC01-WC05 Pubmed
3. Larumbe A. et al. Liquen amiloide con buena respuesta a corticoterapia tópica. Actas Dermosifiliogr 2003;94(6):423-4. FreeTextLink
4. Gómez M. et al. Acral Papular Mucinosis. An Bras Dermatol. 2016;91(5):S111-3 Pubmed
5. Ankad B. et al. Hypertrophic Lichen Planus versus prurigo nodularis: a dermoscopic perspective. Dermatol Pract Concept. 2016;6(2):3 Pubmed
CASO 80
15 de julio
Dra. Mónica Salazar. Residente de Dermatopatología. Programa Dermatopatología
Dr. Andres Flórez. Coordinador Programa Dermatopatología. Universidad CES. Medellín, Colombia.
Imágen clínica cortesía Dra. Carolina Velázquez. Dermatóloga Universidad CES. Medellín, Colombia
Hombre adulto con lesión crateriforme en antebrazo.
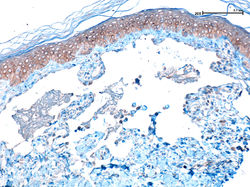
 |  F1Panorámica. Lesión exoendoftítica queratósica. | 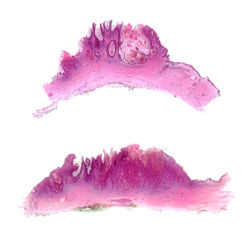 F2Panorámica. Mayor detalle. |
|---|---|---|
 F3Lesión exoendofítica queratósica (arriba). Queratinocitos grandes con citoplasma amplio (abajo) | 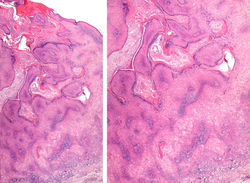 F4 |  F8 |
 F5Mayor detalle, queratinocitos con citoplasma claro, apariencia vitrea, sin atipia. |  F7Mayor detalle, queratinocitos con citoplasma claro, apariencia vitrea, sin atipia. |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Esporotricosis
B. Carcinoma escamocelular ulcerado
C. Queratoacantoma
D. Carcinoma escamocelular de origen folicular
Respuestas
Queratoacantoma María Janeth Vargas
Queratoacantoma Griselda de Anda
Esporotricosis Malena Ortiz G.
Queratoacantoma Anónimo
Queratoacantoma Beatriz di Martino
Queratoacantoma Francisco Chávez R.
Queratoacantoma Jesús Cuevas
Ca. Esca. Folicular Olga Gusova
Queratoacantoma José Luis González
Ca. Esca. Folicular Juan M. González
Queratoacantoma Márcio Martins Lobo
Queratoacantoma Guillermo Ramos R.
Ca. Esca. Folicular Esther Mariela E.
Ca. Esca. Folicular Carlos Cortés Caballero
Queratoacantoma Clara Jaramillo
Queratoacantoma Anónimo
Queratoacantoma Marly Saads Fonseca
Respuesta
C. Queratoacantoma (correcta):
Tumor de superficies expuestas y rápido crecimiento. Se piensa que se origina del folículo piloso (1). La forma más común es la variante esporádica y solitaria que inicia como una pápula y progresa a un nódulo umbilicado en forma de cúpula con tapón hiperqueratósico central (1). Las características histopatológicas son diferentes dependiendo del estadio de la lesión. Se reconocen cuatro etapas: proliferativa, bien desarrollada, en regresión y regresión completa (2). En las dos primeras etapas se pueden reconocer una arquitectura simétrica, crateriforme, exoendofítica (F1,2), con estructuras infundibulares dilatadas contiguas y un tapón queratósico central (F3,4). Además de formación de labios epiteliales sobresalientes a los lados de la lesión y la presencia de lóbulos neoplásicos con diferenciación ístmica (proliferación de células grandes color rosa pálido con apariencia vítrea, sin atipia) (F5-8). En las últimas etapas puede ser difícil su reconocimiento por fenómenos de regresión. Recientemente se ha utilizado CK17 y Ki67 para diferenciarlo del carcinoma escamocelular(3).
A.Esporotricosis (incorrecto): múltiples granulomas supurativos, a veces con cuerpos asteroides (fenómeno de Splendore-Hoeppli).
B.Carcinoma escamocelular ulcerado (incorrecto): desorganización completa de la arquitectura epidérmica, pérdida de maduración y falta de polaridad celular. Marcado pleomorfismo displasia, mitosis atípicas y patrón de crecimiento infiltrativo.
D.Carcinoma escamocelular de origen folicular (incorrecto): muestra agregados neoplásicos de carcinoma escamocelular expandiéndose por el infundíbulo folicular con invasión a la dermis profunda.
Bibliografía:
1. Kwiek B., Schwartz R.,Warsaw P. Keratoacanthoma (KA): An update and review. J Am Acad Dermatol. 2016;74:1220-3. PubMed
2. Ogita A., Ansai SI., Misago N. Histopathological diagnosis of epithelial crateriform tumors: Keratoacanthoma and other epithelial crateriform tumors. J Dermatol. 2016;43:1321-1331. PubMed
3. Leblebici C., Pasaoglu E., Kelten C., Darakci S., Dursun N. Cytokeratin 17 and Ki-67: Immunohistochemical markers for the differential diagnosis of keratoacanthoma and squamous cell carcinoma. Oncol Lett. 2017;13:2539-48. PubMed
CASO 79
30 de junio
Dra. Liza Arias. Residente de Dermatopatología. Universidad CES. Medellin. Colombia.
Mujer. 36 años. Lesión solitaria en dorso, asintomática, violácea con halo eritematoso. La lesión cambia con el tiempo.
Respuestas de los participantes comienzan a aparecer el 10 de Julio. Solución y cierre de caso 15 de Julio 2018
 F1Placa violacea con halo eritematoso |  F2 |  F3 |
|---|---|---|
 F4 |  F5 |  F6 |
 F7 |  CD 31 |  F9D2-40 |
 HV 8 |  F8 |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Hemangioma retiforme
B. Sarcoma de Kaposi
C. Linfangioma progresivo
D. Hemangioma en tachuela
Respuestas
Linfangioma progresivo Elías Del Cóndor
Hemangioma en tachuela Julia Mesa
Linfangioma progresivo Diana Verónica Posso
Hemangioma en tachuela María Janeth Vargas
Linfangioma progresivo Giselle Romero Caimi
Hemangioma en tachuela José Luis González
Hemangioma en tachuela Beatriz de Martino
Hemangioma en tachuela Francisco Chavez Rojas
Linfangioma progresivo Sabrina V. Martínez H
Hemangioma en tachuela Marcela Gómez
Linfangioma progresivo Griselda de Anda
Hemangioma en tachuela Melissa Quintero Picón
Linfangioma progresivo Julieta Corral
Linfangioma progresivo Anónimo
Linfangioma progresivo Malena Ortiz Galarza
Linfangioma progresivo Marielos Posada
Linfangioma progresivo Dr K
Linfangioma progresivo Jairo Sandoval
Linfangioma progresivo Gerardo Prada
Hemangioma en tachuela Guillermo Ramos Rodríguez
Linfangioma progresivo María del Consuelo Gómez Y.
Hemangioma en tachuela Erika Blancas García
Respuesta
D. Hemangioma en tachuela (correcta): Clínicamente en la fase aguda se presenta como una pápula eritematoviolácea rodeada por halo hemorrágico, lesión targetoide o en “diana” (F1). Posteriormente, el halo desaparece dejando la pápula central. Más frecuentes en el tronco y extremidades, asociados a trauma. En fases iniciales la dermis papilar muestra vasos alineados, revestidos por células endoteliales prominentes, con proyecciones intraluminales tipo “tachuela” y de aspecto epitelioide(F2-6), en la dermis profunda los espacios vasculares colapsados y angulados disecan los haces de colágeno y rodean los anexos. Hay extravasación de eritrocitos, infiltrado inflamatorio linfocitario y ocasionales trombos de fibrina. En las etapas tardías hay depósito extenso de hemosiderina, fibrosis y los vasos se encuentran colapsados. La tinción de CD31 es positiva (marcador pan-endotelial)(F8), D2-40 ayuda a confirmar la diferenciación linfática(F9), lo que hace que el diagnóstico diferencial con lesiones de origen linfático sea difícil. La tinción para HHV-8 es siempre negativa. (F10)(1).
A. Hemangioendotelioma retiforme (incorrecto): Neoplasia de malignidad intermedia. Placas o nódulos exofíticos. Histológicamente se observa un patrón vascular que semeja los ¨rete testis¨ (2).
B. Sarcoma de Kaposi (SK) (incorrecto): Histológicamente el diagnóstico diferencial con esta entidad se realiza en la fase de placa donde se observan luces vasculares que disecan la dermis. El SK siempre es HHV-8 positivo(3).
D. Linfangioma progresivo (incorrecto): Esta entidad es muy semejante histológicamente y el diagnóstico diferencial puede ser difícil, sin embrago en la clínica no presentan la imagen en "diana" clásica del hemangioma targetoide. Las luces vasculares están “vacias”, son irregulares y se disponen de forma horizontal. Afecta la dermis superficial y profunda, no siendo infrecuente su extensión al tejido celular subcutáneo y el infiltrado inflamatorio es mínimo(4).
Bibiliografia
1. Kakizaki P., Mangueira D., Castelo S., et al. Targetoid hemosiderotic hemangioma-Case report. An Bras Dermatol. 2014;89(6):956-9 PubMed
2. Zarian H., Salmasco R., Alaibac M. Retiform hemangioendothelioma of the lower limbs: a case-based review. Clinical Dermatology 2015; 3(4):123-125 FreetextlinK
3. Radu O., Pantanowitz L. Kaposi Sarcoma. Arch Pathol Lab Med. 2013; (137):289-294 Freetextlink
4. Messeguer F., Sanmartín O., Martorell-Calatayud A. Acquired Progressive Lymphangioma (Benign Lymphangioendothelioma). Actas Dermosifiliog 2010; 9(101):792-7 PubMed