
CASO 119
13 de diciembre
Dr. Reinhard Rodríguez y Dra. Vanessa García Valencia. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Hombre, 30 años, múltiples pápulas asintomáticas, color piel, de 0,4 cc en el pecho. Sin antecedentes familiares.
¿Cuál cree usted es el diagnóstico más probable?
A. Esteatocistoma múltiple
B. Nevus del folículo piloso
C. Siringomas
D. Quiste velloso eruptivo
Solución y cierre del caso 23 de diciembre
CASO 112 (1ra y 2da parte)
3 de Mayo
Dra. Camila Montoya. Patóloga-Dermatopatóloga. Asunción, Paraguay. Dr. Rodrigo Restrepo. Patólogo-Dermatopatólogo. Profesor Programa de Especialización en Dermatopatología. Universidad CES. Medellín, Colombia.
Caso cortesía Dra. Ana María Aristizabal. Dermatológa. Profesora Univerdidad CES, Mdlln, Colombia.
Niña, 13 años. Áreas alopécicas fronto temporo parietales. Se tomaron dos grupos de biopsias en momentos diferentes (F1 primero; F2, F3 y F4 meses después)
Con los hallazgos histológicos ¿cuál cree es el diagnóstico más probable?
A. Alopecia androgénica
B. Alopecia frontal fibrosante
C. Alopecia areata en patrón ofiásico
D. Alopecia por tracción
Fotos clínicas 8 de mayo
Solución y cierre del caso 18 de mayo
 F1 |  F2 |  F3 |
|---|---|---|
 F4 |  F5 |
CASO 120
18 de enero
Dr. Reinhard Rodríguez y Dr. Javier Hernández. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Caso y fotos clínicas cortesía Dra. Carolina Velásquez. Profesora Dermatología Universidad CES. Mdlln, Col
Paciente masculino de 85 años quien presenta mácula hiperpigmentada en la frente.
¿Cuál cree usted es el diagnóstico más probable?
A. Queratosis actínica pigmentada
B. Lentigo solar
C. Nevus displásico
D. Lentigo maligno
Solución y cierre del caso 30 de enero
CASO 119
13 de diciembre
Dr. Reinhard Rodríguez y Dra. Vanessa García Valencia. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Hombre, 30 años, múltiples pápulas asintomáticas, color piel, de 0,4 cc en el pecho. Sin antecedentes familiares.
¿Cuál cree usted es el diagnóstico más probable?
A. Esteatocistoma múltiple
B. Nevus del folículo piloso
C. Siringomas
D. Quiste velloso eruptivo
Solución y cierre del caso 23 de diciembre
Si no te has inscrito, para opinar debes unirte a nuestra lista de correo AQUí
ESTACIÓN PIEL
Programa de Especialización en Dermatopatología
Universidad CES
Casos 2018 Enero - Junio
CASO 78
15 de junio
Dra. Liza Arias. Residente de Dermatopatología. Caso cortesía Dra. Ana Cristina Ruiz. Profesora Dermatopatología. Universidad CES. Med- Col.
Mujer 61 A. Pápula violácea localizada en cuello, región supracavicular izquierda. Un año de evolución. Secreta material acuoso.
Respuestas de los participantes comienzan a aparecer el 18 de Junio. Solución y cierre de caso 30 de Junio 2018
 F1Panorámica. Epidermis hiperplásica con presencia de estructuras glandulares en dermis reticular |  F2Presencia de estructuras ductales dilatadas que semejan ductos ecrinos |  F3Estructuras elongadas en la dermis con epitelio escamoso y mucinoso |
|---|---|---|
 F4Estructuras ductales revestidos por epitelio columnar mucinoso y escamoso |  F5Mayor detalle de las células columnares mucinosas sin atipia |  F6Detalle del epitelio escamoso en relación con las células columnares |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Glándula salival ectópica
B. Siringometaplasia mucinosa
C. Metaplasia adenomatosa ecrina
D. Mucinosis ductal ecrina
Respuestas
Siringometaplasia mucinosa ecrina Beatriz de Martino
Siringometaplasia mucinosa ecrina Malena Ortiz
Siringometaplasia mucinosa ecrina Carmen López Acosta
Mucinosis ductal ecrina Melissa Quintero Picón
Glándula salival ectópica Juan David Rosales
Metaplasia adenomatosa ecrina Ismery Cabello
Siringometaplasia mucinosa ecrina Francisco Chávez Rojas
Siringometaplasia mucinosa ecrina María del Consuelo Gómez
Siringometaplasia mucinosa ecrina Diego Díaz
Siringometaplasia mucinosa ecrina Carlos Cortés Caballero
Siringometaplasia mucinosa ecrina Susan Peralta Montes
Glándula salival ectópica Sandra Quijano Moreno
Siringometaplasia mucinosa ecrina Clara Jaramillo
Respuestas
B. Siringometaplasia mucinosa (correcto):
Se caracteriza por una depresión epidérmica superficial y por debajo de ella la presencia de estructuras elongadas parecidas a ductos ecrinos (F1-F3) que están revestidos por epitelio escamoso, células columnares con mucina y/o células caliciformes (1) (F4-F6). Clínicamente se presentan como lesiones solitarias, en ocasiones con fístula central que drena material claro. Su localización es más frecuente en sitios acrales, pero puede observarse en cualquier otra parte del cuerpo como en nuestro caso (región cervical). Se cree que es un proceso reactivo y no neoplásico. (2)
A.Glándula salival ectópica (incorrecto): Más frecuentemente en cabeza y cuello. Puede ser congénita o secundaria a traumas, cirugía o malignidades. Clínicamente puede observarse como una depresión que drena material mucinoso. Histológicamente son glándulas conductos salivales normales. (3)
C.Metaplasia adenomatosa ecrina (incorrecto): Glándulas alineadas con/sin epitelio cuboidal cribriforme que puede estar asociada a múltiples neoplasias malignas y benignas (carcinoma basoceluar y escamocelular, espiroadenomas-cilindromas, lesiones melanocíticas, etc) (4) D.Mucinosis ductal ecrina (incorrecto): acumulación de mucina y expansión de las capas externas de la porción intradérmica del ducto ecrino y el acrosiringio (4). Clínicamente es una erupción papular eritematosa y prurítica que involucra tronco y extremidades, siendo más frecuente en pacientes VIH. (1,2)
Bibliografía
-
Abbas O., Bhawan J. Syringometaplasia: variants and underlying mechanisms. International Journal of Dermatology. (2016) 55;142-145. PubMed
-
Mckee P, Calonje E, Thomas B, Alexander L. Cutaneus Cyst. En McKee’s pathology of the skin. Fourth edi. Philadelphia: Elsevier; 2012. p. 1671-1687.
-
Yeh T., Tseng S., Weng C., et al. Congenital ectopic fistula of a minor salivary gland. Journal of Pediatric Surgery (2011)46;2187-2189 PubMed
-
Patterson JW. Disease of cutaneous appendages. En Weedon’s Skin Pathology. 4th ed. China: Elsevier; 2016. p. 458-507.
CASO 77
1 de junio
Dra. Mónica Salazar. Residente de Dermatopatología. Universidad CES. Medellín- Colombia.
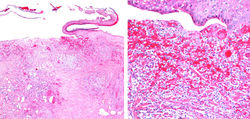
Sexo Femenino. 46 años. Presenta múltiples ulceras dolorosas en miembros inferiores. Varios meses de evolución. No conocida portadora de enfermedad de base. Caso Clínico cortesía del Dr. Ernesto Peña. Dermatólogo.
Respuestas de los participantes comienzan a aparecer el 10 de Junio. Solución y cierre de caso 15 de Junio 2018
 |  |  |
|---|---|---|
 |  |  |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Atrofia blanca
B. Crioglobulinemia
C. PAN
D. Pioderma Gangrenoso
Respuestas
Pioderma gangrenoso Fernando Brenner
PAN Sabrina Herrera
PAN Sandra Viviana Martínez
Pioderma gangrenoso Beatriz de Martino
PAN Juan David Ruiz
Pioderma gangrenoso Márcio Martín Lobos Jardim
PAN Francisco Chávez R.
Pioderma gangrenoso Anónimo
Pioderma gangrenoso Jose David Rosales
Pioderma gangrenoso Marly Saads
Crioglobulinemia Maximiliano Ruiz
Pioderma gangrenoso Marly Fonseca
Pioderma gangrenoso Angela Seidel
Pioderma gangrenoso Anónimo
Pioderma gangrenoso Ismery Cabello
Pioderma gangrenoso Diego Díaz
Pioderma gangrenoso María del Consuelo Gómez
Crioglobulinemia Melissa Quintero Picón
PAN Carlos Cortés Caballero
RESPUESTA
Pioderma gangrenoso (PG)
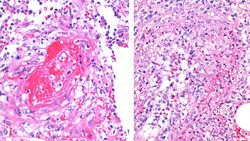
El PG es una enfermedad inflamatoria de etiología desconocida, considerada dentro del grupo de las dermatosis neutrofílicas. Es más común en mujeres y el 50% de los casos se asocia a enfermedades sistémicas. Clínicamente existen cinco subtipos y el más común es el PG ulcerativo, cuya ulcera clásica es de gran tamaño con base necrótica, bordes violáceos, socavados y sobreelevados, rodeada por un halo eritematoso (F1).Pueden ser úlceras únicas o múltiples y son más frecuentes en miembros inferiores. Los hallazgos histológicos son inespecíficos. Las lesiones se caracterizan por presentar un infiltrado inflamatorio dérmico mixto con predominio de neutrófilos (F2-4). En ocasiones asociado a vasculitis leucocitoclástica y linfocitaria, en la base de la úlcera (F5-6). El diagnóstico clínico e histológico se hace por exclusión, ya que como se mencionó, los hallazgos no son específicos.
A. Atrofia blanca (Incorrecto): Proliferación vascular asociado a hialinización de la pared de los vasos, leve o ausente infiltrado inflamatorio, hemorragia y hemosiderina.
B. Crioglobulinemia (Incorrecto): histológicamente pueden presentar vasculitis leucocitoclástica y trombos hialinos intravasculares.
C. Poliarteritis nodosa (Incorrecto):Clínicamente puede presentar nódulos y úlceras, asociados a vasculitis leucocitoclástica necrotizante de vasos de mediano calibre.
Bibliografia:
1. Ashchyan HJ, Nelson CA, Stephen S, James WD, et al. Neutrophilic dermatoses. Part II. Pyoderma gangrenosum and other bowel and arthritis associated neutrophilic dermatoses. J Am Acad Dermatol. 2018 Apr 10. doi: 10.1016/j.jaad.2017.11.063. [Epub ahead of print]. PubMed
2. Schmieder SJ, Krishnamurthy K. Pyoderma Gangrenosum. [Updated 2018 Feb 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. PubMed, Free Text Link
3. Maverakis E, Shinkai K, Fiorentino D, et al. Diagnostic Criteria of Ulcerative Pyoderma Gangrenosum: A Delphi Consensus of International Experts. JAMA Dermatol. 2018 Apr 1;154(4):461-466. PubMed
CASO 76
15 de mayo
Dra. Yoled Carolina Vizcaíno López. Residente de patología. Universidad de Cartagena - Colombia.
Sexo masculino. 23 años. Presenta numerosas pápulas eritematosas dispuestas a lo largo del tronco y porción proximal de extremidades superiores.
Caso clínico cortesía Dra Luz Marina Gómez Vargas. Profesora Dermatología. Medicina. Universidad Pontificia Bolivariana.
Respuestas de los participantes comienzan a aparecer el 20 de Mayo. Solución y cierre de caso 31 de Mayo 2018
 F1 |  F2 |  F3 |
|---|---|---|
 F4 |  F5 |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Miliaria cristalina
B. Dermatitis de contacto alérgica
C. Picadura por atrópodos
D. Miliaria profunda
Respuestas
Miliaria profunda Beatriz de Martino
Miliaria profunda Agustin Estofan
Miliaria profunda Dante Villamil
Miliaria cristalina Elías del Condor
Miliaria cristalina Beatriz Sierra
Miliaria cristalina Marcela Gómez
Miliaria cristalina Juan C. Bonilla J.
Miliaria cristalina Jairo Sandoval
Miliaria cristalina Francisco Chávez R.
Miliaria profunda José David Rosales
Miliaria profunda Carlos Ronald Martínez
Miliaria profunda Guisellda de Anda
Miliaria cristalina Carlos Cortés
Miliaria cristalina Anónimo
Miliaria profunda Erika Blancas Garcia
Miliaria profunda Diego Díaz
Miliaria profunda Clara Jaramillo
Miliaria profunda Márcio Martíns Lobo J
Miliaria cristalina Melissa Quintero Picón
Miliaria profunda Marbel Karina Corzo
RESPUESTA:
D. Miliaria profunda (correcto):
Es un desorden inflamatorio caracterizado por el bloqueo de los ductos sudoríparos. Hay diferentes tipos: cristalina, rubra y profunda, según el nivel del compromiso del ducto sudoríparo y/o acrosiringio. La miliaria profunda es la forma menos común. Es más frecuente en adultos, en general después de eventos repetidos de miliaria rubra. Clínicamente son pápulas eritematosas, rememorando la “piel de gallina” (1)(F1). En la miliria profunda el compromiso es del ducto ecrino y ocurre en la unión dermoepidérmica. Histológicamente está asociada con infiltrado linfocitario dérmico y espongiosis del ducto en la unión dermoepidérmica (2)(F2-4).
A. Miliaria cristalina (incorrecto): obstrucción superficial del ducto a nivel del estrato córneo, clínicamente son vesículas claras de 1-2 mm. Histológicamente presenta una vesícula dentro o directamente debajo del estrato córneo(1). En este caso se observó asociación ocasional histológica con miliaria cristalina.
B. Dermatitis de contacto alérgica (incorrecto): En los estadios iniciales presenta espongiosis, seguida por la formación de vesículas espongióticas en forma de copa, localizadas en diferentes niveles de la epidermis. Sin embargo, dicha espongiosis no se encuentra en relación con los ductos sudoríparos. La dermis superior contiene un infiltrado usualmente con eosinófilos(1).
C. Picadura por artrópodos (incorrecto): se caracteriza por un infiltrado mixto en forma de cuña. Puede observarse espongiosis, ocasionalmente con formación de vesículas (2), sin embargo, ésta no se halla centrada a nivel del acrosiringio.
Bibliografía
1. Patterson JW. The spongiotic reaction pattern. Weedon’s Skin Pathology. 4th ed. China: Elsevier; 2016. p. [108-109].
2. Calonje E, Brenn T, Lazar A, McKee PH. Spongiotic, psoriasiform and pustular dermatoses. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. China: Elsevier; 2012. p. [199-200].
CASO 75
30 de Abril 2018
Dra. Camila Montoya. Residente de Dermatopatología. Universidad CES. Medellín - Colombia.
Mujer de 59 años, lesión dolorosa en pulgar derecho de varios meses de evolución. Múltiples tratamientos.
Caso clínico cortesía Dra Luz Marina Gómez Vargas. Profesora Dermatología. Medicina. Universidad Pontificia Bolivariana.
Respuestas de los participantes comienzan a aparecer el 7 de Mayo. Solución y cierre de caso 15 de Mayo 2018
 |  |  |
|---|---|---|
 |  |  |
 |  |  |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Granuloma piógeno
B. Queratoacantoma ungular
C. Carcinoma escamoso periungular
D. Melanoma ungular
Respuestas
Melanoma ungular Beatriz de Martino
Melanoma ungular Marcela Gómez
Melanoma ungular Jeffrey Silverman
Melanoma ungular Juan Carlos Bonilla
Melanoma ungular Ramiro Pinedo
Granuloma piógeno Agustín Estofan
Melanoma ungular Esther Mariela Estrada
Melanoma ungular Paola María Chávez T.
Melanoma ungular Jesús Cuevas
Granuloma piógeno Mario Lima
Melanoma ungular Angela Seidel
Melanoma ungular Natalia Coras
Melanoma ungular Carmen López
Melanoma ungular José David Rosales
Melanoma ungular Andrés Campuzano
Melanoma ungular Anónimo
Ca. Esca. periungular Melissa Quintero
Melanoma ungular Bryan Alvarez
Melanoma ungular Maximiliano Ruiz
Melanoma ungular Francisco Chávez R.
Melanoma ungular Julieta Corral
Melanoma ungular Víctor Delgado
Melanoma ungular Ismery Cabello
Melanoma ungular Elías Del Cóndor
Melanoma ungular Gisselle Rey
Melanoma ungular Carlos Cortés
Melanoma ungular Gonzálo de Toro
Respuesta
D. Melanoma ungular (amelanótico) (correcto):
Es una neoplasia rara. Se presentan con mayor frecuencia en la raza negra, en el primer dedo del pie o el pulgar(1). El peor pronóstico se asocia a la detección tardía, como en este caso (F1-2).
Histológicamente existe infiltración de melanocitos atípicos en la onicodermis(1)(F3-4). El componente epitelial muestra un patrón lentiginoso con diseminación pagetoide (F5-6). Los hallazgos histológicos clásicos de otras localizaciones pueden estar ausentes en el aparato ungular y algunos parámetros como el Breslow y el nivel de Clark pueden ser inexactos y difíciles de evaluar debido a las particularidades histológicas y anatómicas de la región(1). Los melanocitos ungulares son MELAN–A, HMB-45 positivos(F7-8), la proteína S100 es mucho menos sensible (2).
A. Granuloma piógeno (incorrecto): tumor vascular benigno, compromete los pliegues o el lecho ungular. Sangran fácilmente e histológicamente es muy diferente del melanoma amelanótico al no observarse atipia melanocítica(3)
B. Queratoacantoma ungular (incorrecta): comprometen con más frecuencia el lecho. Histológicamente son lesiones epiteliales en forma de cráter con material queratínico abundante y gran capacidad destructora.
C. Carcinoma escamoso periungular (incorrecta): es el tumor maligno más frecuente de la región ungular, son lesiones verrucosas, pueden comprometer los pliegues laterales y muestran atipia de queratinocitos(4).
Biografía:
1. Tan K-B, et al. Subungual melanoma: a study of 124 cases highlighting features of early lesions, potential pitfalls in diagnosis, and guidelines for histologic reporting. Am J Surg Pathol. 2007;31(12):1902-12. PubMed
2. Theunis A, et al. Immunohistochemical Study of 40 Cases of Longitudinal Melanonychia. Am J Dermatopathol. 2011;33(1):27-34. PubMed
3. Piraccini BM, et al. Periungual and subungual pyogenic granuloma. Br J Dermatol. 2010;163(5):941-53. PubMed
4. Lecerf P, et al. Retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69(2).PubMed
CASO 74
16 de Abril 2018
Dra. Liza Arias. Residente de Dermatopatología. Universidad CES. Medellín - Colombia.
Hombre con lesión en punta nasal. Varios meses de evolución . Asintomático.
Respuestas de los participantes comienzan a aparecer el 22 abril. Solución y cierre de caso 30 de abril 2018
 F1.Nódulo eritematoso en punta nasal |  F2Aspecto macroscópico de la lesión: Nódulo sólido blanquecinos con apariencia cartilaginosa |  F3Panorámica, tumor dérmico circunscrito |
|---|---|---|
 F4Mayor detalle, componente epitelial con patrón difuso(Izquierda) formación de ductos (derecha) |  F5Cordones y ductos revestidos por células cuboidales |  F6Calcificación |
 S100, en las células de la basales externas de las estructuras tubulares |  F8CEA, positivo en la porción luminal de los ductos |  F9Alcian blue positivo para el estroma mixoide |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Condroma cutáneo
B. Neurofibroma mixoide
C. Tumor mixto apocrino
D. Tumor mixto cutáneo
Respuestas
Tumor mixto cutáneo Beatriz di Martino
Tumor mixto cutáneo María Janeth Vargas
Tumor mixto apocrino Marcela Gómez B.
Tumor mixto cutáneo Ramiro Pinedo
Tumor mixto cutáneo Marcela Saeb Lima
Tumor mixto apocrino Carmen López
Tumor mixto cutáneo Sandra V. Martínez H.
Tumor mixto cutáneo Sabrina Herrera
Tumor mixto cutáneo Juan Manuel González
Tumor mixto apocrino Melissa Quintero P.
Tumor mixto apocrino Anónimo
Tumor mixto cutáneo Luis F. Cárdenas
Tumor mixto apocrino Gerardo Prada
Tumor mixto cutáneo Anónimo
Neurofibroma Mixoide Agustin Estofan
Tumor mixto cutáneo Griselda de Anda
Tumor mixto apocrino Ismery Cabello
Tumor mixto cutáneo Sabrina Herrera
Tumor mixto apocrino Marielos Posada
Tumor mixto apocrino Carlos Ronald Martínez
Tumor mixto cutáneo Esther Mariela Estrada
Tumor mixto apocrino Clara Jaramillo
Tumor mixto apocrino Francisco Chávez R
Neurofibroma Mixoide Jose David Rosales
Tumor mixto cutáneo Carlos Cortés Caballero
Tumor mixto apocrino Erika Blancas García
Tumor mixto cutáneo Guillermo Ramos
Tumor mixto cutáneo María del Consuelo Gómez
Respuesta
C. Tumor mixto cutáneo (correcta).
Es una lesión de crecimiento lento, indolente y benigna que se presenta en cabeza y cuello en personas de edad avanzada (F1-2). Son tumores dérmicos multilobulados, pueden comprometer el tejido celular subcutáneo(F3), forman estructuras tubuloalveolares revestida por una o dos capas de células cuboidales, dispuestas en cordones o nidos (F4-6) asociado a una matriz de apariencia condroide que se tiñe con ltinción PAS y Alcian blue (F9). El epitelio ductal es positivo para citoqueratinas y CEA (en la porción luminal)(F8) mientras que las capas externas expresan vimentina y S100. (F7)(1)
A.Condroma cutáneo: (incorrecta):
Ocurren usualmente en manos y pies. Tumor intradérmico o del tejido celular subcutáneo, bien circunscrito formado por cartílago hialino maduro, que puede presentar cambio mixoide, calcificación u osificación(2).
B. Neurofibroma mixoide (incorrecta):
Es una variante del neurofibroma convencional con un depósito de mucina en el estroma(3).
C. Tumor mixto apocrino (incorrecta):
Neoplasia benigna con un componente epitelial que presenta diferenciación tubular con secreción por decapitación, algunos autores consideran que esta entidad hace parte del espectro de los tumores mixtos cutáneos(4).
Bibliografia:
-
Villalón G., et al. Siringoma condroide: revisión clínica e histológica de ocho casos. Actas Dermosifiliogr 2006; 97:573-7 FreeTextLink
-
Yamaguchi Y, et al. A case of condroma cutis showing callus-like appearance. J Eur Acad Dermatol Venereol. 2017 Jan;31(1): e37-e38. PubMed
-
Kane P, et al. Solitary mixoid neurofibroma of the palm. J Hand Microsurg. 2015 Dec;7(2):330-1. PubMed
-
Vicioso L., et al. Cutaneous mixed tumor with lipomatous stroma. J Cutan Pathol 2006:33(2):35-38 . PubMed
CASO 73
1 de Abril 2018
Dra. Mónica Salazar. Residente de Dermatopatología. Universidad CES. Medellín - Colombia.
Hombre 23 años de edad. Presenta desde hace cuatro años placa eccematosa en el lado izquierdo de la nariz, asintomática. Múltiples tratamientos sin mejoría.
Respuesta 15 de abril 2018
 F1. Lesión original |  F2. Aspecto antes de la resección |  F3. Biopsia |
|---|---|---|
 F4. Biopsia |  F5. Biopsia |  F6. Resección quirúrgica |
 F7. Resección quirúrgica |  F8. Resección quirúrgica |  F9. Antígeno carcinoembrionario |
 F10. Antígeno carcinoembrionario |
Nota: Fotos 3 a 5 biopsia por Punch. Fotos 6 a 11 : resección / ampliación
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Tricoadenoma
B. Tricoepitelioma
C. Carcinoma anexial microquístico
D. Esporotricosis
Respuestas
Ca. Anex. microquístico Sabrina Herrera
Ca. Anex. microquístico Ramiro Pinedo
Tricoepitelioma Marly Saads
Ca. Anex. microquístico Luis F. Cárdenas C.
Ca. Anex. microquístico Alvaro Peynado
Ca. Anex. microquístico Francisco Chávez
Tricoepitelioma Angela Seydel
Tricoepitelioma Mario A. Melo
Ca. Anex. microquístico Carlos Cortés C.
Ca. Anex. microquístico Ismery Cabello
Ca. Anex. microquístico Angela Suárez
Ca. Anex. microquístico Gerardo Prada
Ca. Anex. microquístico Esther Mariela E.
Ca. Anex. microquístico Marcela Saeb Lima
Ca. Anex. microquístico Anónimo
Ca. Anex. microquístico Diana Castillo
Ca. Anex. microquístico María del Consuelo Gómez
Ca. Anex. microquístico Clara Jaramillo
Ca. Anex. microquístico Marcela Gómez Barrera
Respuesta:
C. Carcinoma anexial microquístico (CAM)
El CAM es un neoplasia cutánea maligna localmente agresiva, con diferenciación apocrina, folicular y sebácea(1), localizada preferentemente en cabeza y cuello en individuos entre 44 y 64 años(2). De etiología desconocida, se asocia a factores predisponentes como radioterapia, exposición a radiación ultravioleta e inmunosupresión. Clínicamente son nódulos o placas induradas de crecimiento lento (F1-2)(3).
Histológicamente presenta compromiso superficial y profundo. El componente superficial está compuesto por numerosos queratoquistes, pequeñas islas o cordones de epitelio basaloide y escamoso con diferenciación ductal. Pueden simular siringomas, tricoepiteliomas
y carcinoma basocelular(F3-5). El componente profundo muestra nidos más pequeños y cordones de células sobre un estroma fibroso hialinizado (F6-7)(3). La invasión perineural es frecuente (F8-10). La inmunohistoquímica muestra positividad para CEA en 50% de los casos (F9-10), y negatividad para Ber-EP4. No existen tinciones específicas para esta entidad (2,3). En biopsias superficiales con escasa diferenciación ductal, como este caso (F3-5) se debe estudiar toda la lesión, ya que el compromiso profundo y la invasión perineural solo fueron vistas en la resección completa( F6-11)
A. Tricoadenoma (Incorrecto): Histológicamente presenta quistes con diferenciación ístmica. No hay invasión perineural ni ductos.
B. Tricoepitelioma (Incorrecto): Pápulas únicas o múltiples centrofaciales, muestran nidos de células basaloides con quistes córneos y cuerpos mesenquimales. Sin diferenciación ductal ni invasión perineural.
D. Esporotricosis (Incorrecto): Puede presentarse como placas. Histológicamente es inflamatoria, con infiltrado mixto y granulomas. Los cuerpos asteroides, levaduras en forma de cigarro se pueden observar ocasionalmente. El cultivo confirma el diagnóstico.
Bibliografía:
-
Mamic M, et al: Microcystic adnexal carcinoma-diagnostic criteria and therapeutic methods: case report and review of the literature. Int. J. Oral Maxillofac. Surg. 2018. PubMed
-
Aslam A, Microcystic Adnexal Carcinoma and a Summary of Other Rare Malignant Adnexal Tumours. Curr. Treat. Options in Oncol. 2017; 18:49. PubMed
-
Gordon S, et al. Microcystic Adnexal Carcinoma: A Review of the Literature. Dermatol Surg 2017; 0:1–5. PubMed
CASO 72
15 de marzo 2018
Dra. Jaqueline Cifuentes. Residente de Dermatología. Universidad del Valle. Cali - Colombia.
Hombre 40 años, 6 meses de evolución de placas atróficas en tronco, antecedente de sífilis secundaria hace 4 años. Actual VRDL 1:32 diluciones. Anticaridolipinas IgM 6.3 IgG 12.2
Fotos clínicas cortesía Dra. Luz Marina Gómez V y Angela Londoño, Dermatólogas y profesoras UPB y CES.
Respuesta 1 de abril 2018
 F1Placas atróficas en tronco |  F2Mayor detalle de las placas atróficas |  F3placas atróficas en dorso |
|---|---|---|
 F4Placas atróficas en pliegue axilar y miembro superior |  F5Mayor detalle de las placas, todas de iguales características |  F6Panorámica. epidermis de características normales, ausencia de inflamación dérmica. |
 F7A mayor detalle epidermis con características usuales y ausencia de inflamación. Puede ser interpretado como piel normal en ausencia de datos clínicos. |  F8Colágeno dérmico de aspecto normal |  F9 |
 F10Tinción para fibras elásticas con orceína, disminución de las fibras elásticas en la dermis superficial |  F11Tinción para fibras elásticas con orceína. Fibras elásticas distorsionadas y fragmentadas (elastolisis y elastorexis). |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Morfea en placas
B. Anetodermia cutánea primaria
C. Anetodermia secundaria a sífilis
D. Elastolisis de la dermis media
Respuestas
Morfea en placas Dante villamil
Anetodermia 2ria sífilis María Paula Abelenda
Anetodermia 2ria sífilis Angela Seidel
Anetodermia 2ria sífilis Marcelo Toro A
Elastolisis de la dermis media Melissa Quintero
Anetodermia 2ria sífilis Griselda de Anda
Anetodermia 2ria sífilis Daniela Gómez O
Anetodermia 2ria sífilis Anónimo
Anetodermia 2ria sífilis Andrés F. López R
Anetodermia 2ria sífilis Ismery Cabello
Anetodermia 1ria Marly Saads Fonseca
Anetodermia 2ria sífilis María del Consuelo Gómez
Elastolisis de la dermis media Carlos Cortés caballero
Anetodermia 2ria sífilis Alvaro Peinado. Diferencial REM
Anetodermia 2ria sífilis Naima Rebaiai. Ca peut être une Chéloide.....Morphée en plaque (sclérodermie) vu l'aspect aminci et rectiligne de l’épidermePuede ser una queloide
Anetodermia 2ria sífilis María Janeh Vargas
Anetodermia 2ria sífilis Clara Jaramillo
Respuesta
C. Anetodermia secundaria a sífilis
En los años 90 la sífilis secundaria fue la primera causa de anetodermia, pero en las ultimas décadas han disminuido los reportes de este hallazgo. La anetodermia es una rara entidad benigna de diversa etiología, donde hay disminución o ausencia de fibras elásticas dérmicas. Puede ser idiopática o secundaria a desórdenes tumorales, infecciosos, inflamatorios y autoinmunes, sin tener una patogenia clara. Clínicamente son placas atróficas y laxas, más frecuente en el tronco y extremidades superiores, como en este caso (F1-F5).
En la histopatología los hallazgos pueden ser sutiles e incluso pasar desapercibidos si no se acompañan de datos clínicos, se caracteriza por reducción importante del número de fibras elásticas y discreto infiltrado perivascular superficial linfocítico con o sin presencia de histiocitos (F6-F8). La reducción de fibras elásticas en la dermis se evidencia con las coloraciones especiales (F9-F10). En la epidermis es posible encontrar mezcla de patrón psoriasiforme y liquenoide (2)
A: Morfea en placas (Incorrecto): Se asocia a cambios escleróticos en la piel, sin pérdida de las fibras elásticas(3)
B: Anetodermia cutánea primaria (incorrecto): Se descarta esta posibilidad debido a la relación con las lesiones luéticas previas.
D: Elastolisis de la dermis media (incorrecto): Clínicamente son placas atróficas y se asocia a disminución de las fibras elásticas sin embargo en esta entidad la disminución es focal y en banda en la dermis media(1)
Bibliografía
1. Martínez ME. Mid dermal elastolysis: another dermatological clue to autoimmunity Acta Derm Venereol 2011. PubMed
2. Veasey JV. Anetoderma due to secondary syphilis: Report of two cases and discussion of the histopathological findings. SAGE journal 2017 Dec;28(14):1456 1460 PubMed
3. Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015 Jan-Feb;90(1):62-73 PubMed
CASO 71
1 de marzo 2018
Dra. Alejandra Avila. Residente de Dermatología. UPB. Medellín Colombia.
Hombre, 33 años. Seis meses de evolución de lesión en espalda, asintomático.
Respuesta 15 de marzo 2018
 F1 |  F2 |  F3 |
|---|---|---|
 F4 |  F5 |  F6 |
 F7Azul de Prusia |
Con las características clínicas e histológicas el diagnóstico más probable es:
A. Queratosis liquenoide
B. Liquen aureus
C. Necrobiosis lipoídica
D. Dermatitis liquenoide purpúrica
Respuestas
Liquen aureus Anónimo
D. L. Purpúrica Juan Felipe Pinilla
D. L. Purpúrica Dante Villamil
Q. Liquenoide Melissa Quintero Picón
Liquen aureus Griselda de Anda
D. L. Purpúrica Cibele Pereira
Liquen aureus Helga Sarti
Q. Liquenoide Marco Rosillo Páez
Liquen aureus Marly Saads
Liquen aureus Esther Mariel E.
D. L. Purpúrica Diana de la Cruz
Liquen aureus Clara Jaramillo
D. L. Purpúrica Lucy Bravo
Respuesta
B. Liquen Aureus (LA)
Es la forma menos común de las dermatosis purpúricas . Se presenta como una mácula o placa liquenoide, bien delimitada, rojiza, parduzca o violácea(F1). Generalmente solitaria, asintomática o levemente pruriginosa. Aparece preferentemente en miembros inferiores, pero también en tronco y miembros superiores, como en el caso que presentamos. Es más frecuente en niños y adultos jóvenes varones(1). Su etiología es desconocida
Histopatológicamente, el (LA) se caracteriza por un infiltrado liquenoide variable (F2) de linfocitos e histiocitos, mezclado con eritrocitos extravasados (F3-F5) y depósitos de hemosiderina en la dermis papilar(F6) (2). Sin embargo, se ha descrito otros patrones de reacción inflamatoria, como lo son el granulomatoso y el perivascular e intersticial profundo(3).
El diagnóstico diferencial histopatológico en ocasiones es difícil, ya que puede confundirse con otras dermatosis purpúricas, el liquen estriado, la micosis fungoide purpúrica (3) y la queratosis liquenoide, como sucedió en este caso, por esta razón, la discusión con el dermatólogo ayudó de una manera importante a aclarar el diagnóstico.
En el tratamiento se ha descrito el uso de esteroides tópicos, inhibidores de la calcineurina y ácido ascórbico, todos con resultados limitados(1).
Referencias
1.Torraca PFS, Murback NDN, et al. Segmental lichen aureus: an uncommon presentation. An Bras Dermatol. 2017 Sep-Oct;92(5):704-706.PubMed Central PMCID: PMC5674707. PubMed
2.Zeng YP, Fang K, et al. Lichen aureus: clinicopathological features in a Chinese series. Eur J Dermatol. 2016 Jun 1;26(3):290-4. PubMed PMID: 27101633. PubMed
3.Aung PP, Burns SJ, et al. Lichen aureus: an unusual histopathological presentation: a case report and a review of literature.Am J Dermatopathol 2014; 36: e1-4. PubMed
CASO 70
15 de febrero 2018
Dra. Liza Arias. Residente de Dermatopatología. Universidad CES. Medellín Colombia.
Caso clínico cortesía Dr. Ernesto Peña S.
Mujer 35 años. Múltiples lesiones papulares de consistencia firme. Varios años de evolución.
Respuesta 28 de febrero 2018
 F1Pápulas, firmes, rosadas-amarillentas en rostro. |  F2. TAC de cráneoSe observan en la región anterior múltiples estructuras de densidad ósea, independientes de los huesos faciales |  F3Nódulo bien circunscrito con hueso compacto maduro y tejido adiposo |
|---|---|---|
 F7Mayor detalle de hueso lamelar maduro |  F4Panorámica de nódulo formado por hueso lamelar maduro y centro medular |  F5Hueso compacto maduro con presencia de osteocitos |
 F6Hueso compacto maduro con presencia de osteocitos y reborde de osteblastos |

Con las características clínicas e histológicas suministradas, el diagnóstico más probable es:
A. Sarcoma de Kaposi.
B. Vasculitis
C. Acroangiodermatitis.
D. Dermatosis purpúrica pigmentaria.
Respuestas
Osteoma cutis Clara Jaramillo
Osteoma cutis Jennifer Castillo G.
Osteoma cutis Verónica Posso
Osteoma cutis Joyce B. Forero
Osteoma cutis Anónimo
Osteoma cutis Marcelo Toro A.
Osteoma cutis Gerardo Prada
Osteoma cutis Melissa Quintero P.
Osteoma cutis Esther Mariela Estrada
Osteoma cutis Juan M. González C.
Osteoma cutis Carolina Delgado
Osteoma cutis Cibele Conceição dos Apóstolos Pereira
Osteoma cutis Dante Villamil
Osteoma cutis Andrés Fabian López R
Osteoma cutis Pilar Alemán
Osteoma cutis María del Consuelo Gómez
Osteoma cutis Carlos Cortés
Osteoma cutis Ismery Cabello
Osteoma cutis Juan Felipe Pinilla
Respuesta
D. Osteoma cutis miliar múltiple
Variante de osteoma cutis caracterizado por ser un desorden raro y benigno con formación de focos de tejido óseo dentro de la dermis e hipodermis(1). Más frecuente en mujeres entre la tercera y cuarta década de la vida(2). Se puede clasificar en forma primaria asociada a síndromes genéticos y la forma secundaria que constituye el 80% de los casos y se produce como consecuencia de lesiones previas (acné) y en enfermedades sistémicas(2). Son pápulas normocrómicas y duras con predilección por el rostro (F1). (1) Existen dos teorías en cuanto a su etiología: 1) metaplasia de los fibroblastos y 2) Migración errónea de las células mesenquimales embrionarias hacia la dermis diferenciándose en estructuras osteogénicas(1). La histología evidencia depósito de hueso lamelar el cual se caracteriza por osteocitos en el centro y osteoclastos en la periferia asociado a colágeno mineralizado y condensado (F3-6). El diagnóstico es facilitado por tomografía computarizada la cual muestra elementos independientes de los huesos faciales (F2) (2,3). El tratamiento consiste en la escisión quirúrgica de cada lesión, pero existen múltiples modalidades entre ellas la dermoabrasión, láser Co2 y YAG (2). (ver caso 22)
A. Acné comedogénico (incorrecta): desorden de la unidad pilosebácea que se presenta con comedones abiertos y cerrados(4).
B. Tricoepiteliomas (incorrecta): Tumor benigno de origen folicular formado por nidos de células basaloides(5).
C. Fibrofoliculomas (incorrecta): hamartoma benigno que consiste en un folículo dilatado con múltiples bandas de células epiteliales que emergen del infundíbulo(5).
Bibliografía:
-
Bouraoui S., et al. Miliary osteoma cutis of the face. J Dermatol Case Rep. 2011:5(44) pp:77-81. FreeTextLink
-
Aguinaga F, et al. Miliary osteoma cutis: a case report. Case reports in Dermatological Medicine. 2014. Pp: 1-3. Freetextlink
-
Alhazmi D, et al. Osteoma cutis of the face in CBTC images. Case Reports in Dentistry. 2017. Pp: 1-4 FreeTextLink
-
Ivars M, et al. Trastornos del folículo pilosebáceo: acné y rosácea. Medicine. 2014:11(48);2823-2839. Abstract
-
Singh S,et al. Multiple familial trichoepiteliomas presenting as leonine facies. Indian Dermatol Online J. 2017:8(5);358-360 FreeTextLink
CASO 69
1 de febrero 2018
Dra. Mónica Salazar. Residente de Dermatopatología. Universidad CES. Medellín Colombia.
Hombre, 70 años. Sin antecedentes patológicos de importancia. Presenta desde hace 9 meses placas eritematovioláceas bien delimitadas, asintomáticas, localizadas en piernas.
Respuesta 15 de febrero 2018
 F1Dermis superficial: incremento en el número de capilares y dispuestos en lóbulos |  F2Dermis superficial: mayor detalle |  F3Hiperplasia epidérmica asociada a proliferación de capilares en la dermis con extravasación de eritrocitos y hemosiderina |
|---|---|---|
 F4Mayor detalle: proliferación de capilares con paredes engrosadas |  F5. CD31 |  F6. Azul de prusia |
Con las características clínicas, histológicas y de imágenes suministradas, el diagnóstico más probable es:
A. Acné comedogénico
B. Tricoepiteliomas
C. Fibrofoliculomas
D. Osteoma cutis
Respuestas
Sarcoma de Kaposi Sara Acosta Cajo
Acroangiodermatitis Herberg Eastman
Acroangiodermatitis Verónica Alba
Acroangiodermatitis Dante Villamil
Acroangiodermatitis Juan Felipe Pinilla
Acroangiodermatitis Ana Cristina Ruiz
Acroangiodermatitis Juan M. Gozález
Acroangiodermatitis Marcelo Toro Arias
Sarcoma de Kaposi Carlos Ronald Martínez C
Sarcoma de Kaposi Cibele Conceição dos Apóstolos
Acroangiodermatitis Andrés Fabián López
Sarcoma de Kaposi Roniel Contreras
Acroangiodermatitis Camila Montoya B
Sarcoma de Kaposi Esther Mariela Estrada
Acroangiodermatitis Gerardo Prada
Sarcoma de Kaposi Julieta Corral
Acroangiodermatitis Jesús Cuevas
Sarcoma de Kaposi Erika Blancas García
Acroangiodermatitis Melissa Quintero
Acroangiodermatitis Jaime Arturo Mejía
Sarcoma de Kaposi Carlos Cortés Caballero
Acroangiodermatitis Guillermo Ramos Rodríguez
Acroangiodermatitis Jenifer Castillo
Respuesta
C. Acroangiodermatitis (pseudo Kaposi, enfermedad de Mali).
Es una enfermedad cutánea angioproliferativa benigna, su etiología no está clara, sin embargo se ha relacionado con insuficiencia venosa crónica, malformaciones o fistulas arteriovenosas, extremidades paralíticas, uso de prótesis en pacientes amputados, trombofilias e incluso el uso de fármacos intravenosos(1). Se han descrito diversas variantes como el síndrome de Stewart-Bluefarb (malformación A-V congénita), tipo Mali (asociada a insuficiencia venosa crónica) e incluso purpura del embarazo (Dermatitis ocre de Favre) y en pacientes en hemodiálisis después de la confección de la fistula A-V(2).
Clínicamente se caracteriza por presencia de máculas, pápulas, placas o nódulos violáceos en las extremidades inferiores, sobretodo en tobillos y dorso de pies. La histopatología muestra tanto en la dermis superficial y profunda incremento en el número de capilares (Fig 1-3), con paredes engrosadas (Fig. 4), y extravasación de hematíes(Fig.3), además se observa depósitos de hemosiderina (Fig.3,4 y 6) acompañado de un infiltrado mixto con escasas células plasmáticas. Puede haber trombosis intracapilares. Inmunohistoquimicamente muestra positividad para marcadores endoteliales como CD31 (Fig.5) y CD34 (1).
A. Sarcoma de Kaposi (SK): (Incorrecta) su distinción con acroangiodermatitis puede ser difícil, sin embargo existen ciertos hallazgos que pueden resultar útiles: en el SK se observa signo del promontorio, proliferación de células endoteliales atípicas, proliferación vascular independiente de los vasos existentes e infiltrado perivascular con abundantes células plasmáticas(1)
B. Vasculitis (Incorrecta) : A diferencia de la acroangiodermatitis en la vasculitis existe daño e inflamación de las paredes vasculares.
D. Dermatosis púrpurica pigmentaria (incorrecta): Se observa dilatación capilar con edema endotelial como característica común, a diferencia de la acroangiodermatis que muestra incremento en el número de capilares(2)
Bibliografía:
1. Coban I, Kokenek-Unal T, Alper M. Spontaneous Acroangiodermatitis. Indian J Dermatol. 2015 May-Jun; 60(3): 268–271. PubMed
2. Kumar S, Manchanda K. Acroangiodermatitis (Pseudo-Kaposi sarcoma). Indian Dermatology Online Journal - July-September 2014 - Volume 5 - Issue 3. PubMed
CASO 68
15 enero 2018
Dra. Catalina Cuellar. Dermatopatóloga. Universidad CES. Medellín Colombia.
Mujer. 51 años. 4 meses de evolución de aparición súbita de múltiples lesiones pruriginosas localizadas en brazos y tórax. Lesión similar única en abdomen de 5 años de evolución. Ningún otro hallazgo clínico.
Respuesta 30 de enero 2018
 |  |  |
|---|---|---|
 |  |  |
 Robert Degos (1904–1987)https://commons.wikimedia.org/wiki/File%3ADegos%2C_Robert_Gaston_CIPB1884.jpg |
Con las características clínicas e histológicas suministradas, el diagnóstico más probable es:
A. Enfermeda de Degos
B. Lupus eritematoso cutáneo
C. Esclerosis sistémica
D. Dermatomiositis
Respuestas
Enfermedad de Degos Chester Nimitz
Enfermedad de Degos Kalyani Bambal
Enfermedad de Degos Bibiana Peña
Enfermedad de Degos Jennifer Castillo
Enfermedad de Degos Griselda de Anda
Lupus eritematoso cutáneo Herberg Eastman
Enfermedad de Degos Juan Carlos Garcés
Enfermedad de Degos Jesús Pérez García
Enfermedad de Degos Gerardo prada
Enfermedad de Degos Hernando Marín
Lupus eritematoso cutáneo Carmen López Acosta
Lupus eritematoso cutáneo Mariangela Marques
Dermatomiositis Melissa Quintero Picón
Enfermedad de Degos Ronald Martínez Castellanos
Enfermedad de Degos Marcia Alvarenga Lira
Papulosis atrófica Maligna Antonio Guzmán
Enfermedad de Degos Marly Saads
Lupus eritematoso cutáneo Ismery Cabello
Enfermedad de Degos Esther Mariela Estrada
Enfermedad de Degos Milton Mejía
Enfermedad de Degos María del Consuelo Gómez
Respuesta
A. Enfermedad de Degos (Köhlmeier-Degos)
La papulosis atrófica maligna es una condición poco frecuente. Se manifiesta usualmente entre la tercera y sexta década de la vida (1). Se asocia con un rasgo genético autosómico dominante(1).
El diagnóstico se hace con base en la clínica, que es patognomónica. Son pápulas con centro atrófico con aspecto de porcelana y borde eritematoso con telangiectasias (F1-F2)(1). Afecta con preferencia el tronco y extremidades superiores. Aparecen como pequeñas pápulas eritematosas, cuyo centro se deprime después de algunos días, adquiriendo el aspecto característico descrito (1).
Puede asociarse a compromiso de órganos internos, con infartos múltiples; en estos casos, se denomina como “enfermedad de Degos maligna” por su pronóstico desfavorable, comparado con los casos limitados a la piel, como este que presentamos (1,2). La afectación de órganos internos puede ocurrir simultáneamente o incluso años después de la aparición de las lesiones cutáneas, y usualmente acarrea complicaciones sistémicas serias secundarias a la isquemia(1).
En la histopatología se observa en las lesiones tempranas un infiltrado linfocítico superficial y profundo perivascular (F3-F4), con depósitos de mucina(F6), similar a los hallazgos del lupus cutáneo. Posteriormente se hacen evidentes los cambios en la unión dermoepidérmica, con atrofia de la epidermis y esclerosis de la dermis papilar (F5). En las lesiones tardías, se ve necrosis en cuña del tejido conectivo por oclusión de las arterias dérmicas (1).
La fisiopatología de la papulosis atrófica maligna es controvertida, las teorías que se han propuesto son la vasculitis, coagulopatía o una disfunción primaria de las células endoteliales, sin embargo, ninguna de ellas ha sido demostrada hasta ahora (1).
Bibliografía
1. Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease) – A review. Orphanet J Rare Dis. 2013;8:10 Pubmed
2. Viswanath V, Gada JL, Shah RJ. Degos disease: A murderous menace. Indian J Dermatol 2016;61:572-4 Pubmed