
CASO 119
13 de diciembre
Dr. Reinhard Rodríguez y Dra. Vanessa García Valencia. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Hombre, 30 años, múltiples pápulas asintomáticas, color piel, de 0,4 cc en el pecho. Sin antecedentes familiares.
¿Cuál cree usted es el diagnóstico más probable?
A. Esteatocistoma múltiple
B. Nevus del folículo piloso
C. Siringomas
D. Quiste velloso eruptivo
Solución y cierre del caso 23 de diciembre
CASO 112 (1ra y 2da parte)
3 de Mayo
Dra. Camila Montoya. Patóloga-Dermatopatóloga. Asunción, Paraguay. Dr. Rodrigo Restrepo. Patólogo-Dermatopatólogo. Profesor Programa de Especialización en Dermatopatología. Universidad CES. Medellín, Colombia.
Caso cortesía Dra. Ana María Aristizabal. Dermatológa. Profesora Univerdidad CES, Mdlln, Colombia.
Niña, 13 años. Áreas alopécicas fronto temporo parietales. Se tomaron dos grupos de biopsias en momentos diferentes (F1 primero; F2, F3 y F4 meses después)
Con los hallazgos histológicos ¿cuál cree es el diagnóstico más probable?
A. Alopecia androgénica
B. Alopecia frontal fibrosante
C. Alopecia areata en patrón ofiásico
D. Alopecia por tracción
Fotos clínicas 8 de mayo
Solución y cierre del caso 18 de mayo
 F1 |  F2 |  F3 |
|---|---|---|
 F4 |  F5 |
CASO 120
18 de enero
Dr. Reinhard Rodríguez y Dr. Javier Hernández. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Caso y fotos clínicas cortesía Dra. Carolina Velásquez. Profesora Dermatología Universidad CES. Mdlln, Col
Paciente masculino de 85 años quien presenta mácula hiperpigmentada en la frente.
¿Cuál cree usted es el diagnóstico más probable?
A. Queratosis actínica pigmentada
B. Lentigo solar
C. Nevus displásico
D. Lentigo maligno
Solución y cierre del caso 30 de enero
CASO 119
13 de diciembre
Dr. Reinhard Rodríguez y Dra. Vanessa García Valencia. Residentes Dermatopatología, Universidad CES. Dra. Camila Montoya. Patóloga- Dermatopatóloga. MSPBS. Asunción, Paraguay.
Hombre, 30 años, múltiples pápulas asintomáticas, color piel, de 0,4 cc en el pecho. Sin antecedentes familiares.
¿Cuál cree usted es el diagnóstico más probable?
A. Esteatocistoma múltiple
B. Nevus del folículo piloso
C. Siringomas
D. Quiste velloso eruptivo
Solución y cierre del caso 23 de diciembre
Si no te has inscrito, para opinar debes unirte a nuestra lista de correo AQUí
ESTACIÓN PIEL
Programa de Especialización en Dermatopatología
Universidad CES
Casos 2016 Julio - Diciembre
CASO 46
5 de Diciembre - Último caso de este año
Dr. Andrés Flórez. Profesor de Dermatopatología. Universidad CES. Medellín, Colombia.
Hombre adulto con lesión nodular en la frente. Dolor espontáneo y a la palpación.
Con las características clínicas e histológicas suministradas el diagnóstico más probable es:
a. Espiradenoma
b. Tricofoliculoma
c. Neurofibroma
d. Hidradenoma nodular
e. Linfoma cutáneo
Respuesta 30 de enero 2017
 El diagnóstico clínico es difícil. Se presenta como un tumor solitario y se localiza en cabeza y cuello. Afecta adolescentes y adultos, sin predilección por sexo. |  Su diagnóstico es histológico observadose uno o varios lóbulos de forma oval ubicados en la dermis y/o tejido celular subcutáneo. |  Son generalmente sólidos o con estructuras quísticas y tubulares. |
|---|---|---|
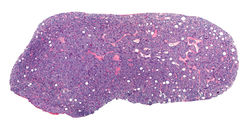 Pueden tener una cápsula fibrosa de tejido conectivo. |  Pueden tener una cápsula fibrosa de tejido conectivo. A veces hay depósito intercelular de material de membrana basal en un patrón en rompecabezas. |  Este patrón es muy similar al observado en los cilindromas, dando lugar a formar mixtas conocidas como espiradenocilindromas. |
 Los lóbulos están constituidos por cordones entrelazados que contienen dos tipos de células epiteliales: unas de núcleo oscuro, basofílicas, que se localizan en la periferia alrededor de los vasos sanguíneos; otras de núcleo grande, pálido y ovoide, con citoplasma abundante que están ubicadas en el centro de los lóbulos. |  Se observan linfocitos en cantidad variable, los cuales se disponen entre las células oscuras y claras o dentro del estroma fibroso que rodea al tumor. | 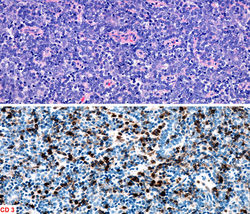 El infiltrado linfoide es CD3 positivo. |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
Espiradenoma María Janeth Vargas
Espiradenoma Elsa Bibiana Peña
Espiradenoma Gerardo Prada
Espiradenoma Mónica Ruiz
Espiradenoma Clara jaramillo
Espiradenoma
El espiradenoma es un tumor benigno de origen glandular cuya histogénesis exacta ha sido muy debatida. Clásicamente se había considerado un tumor ecrino.
El diagnóstico clínico es difícil. Se presenta usualmente como un tumor solitario y asintomático (aunque se ha descrito dolor espontáneo al cambio de presión o temperatura, como en este caso). Se localiza principalmente en cabeza, cuello y tórax, menos frecuente en brazos y piernas. Afecta principalmente adolescentes, adultos jóvenes y ancianos, sin predilección de sexo. Su evolución es crónica y excepcionalmente puede haber transformación maligna. Hay reportes de casos congénitos, múltiples, familiares y relacionados con otros tumores benignos de glándulas sudoríparas, como en el Síndrome de Brooke-Spiegler, asociado además a cilindromas y tricoepiteliomas..
Su diagnóstico es histológico observadose una neoformación constituida por uno o varios lóbulos de forma oval ubicados en la dermis y/o tejido celular subcutáneo, generalmente sólidos o con estructuras quísticas y tubulares que pueden tener una cápsula fibrosa de tejido conectivo. Los lóbulos están constituidos por cordones entrelazados que contienen dos tipos de células epiteliales: unas de núcleo oscuro, basofílicas, que se localizan en la periferia alrededor de los vasos sanguíneos; otras de núcleo grande, pálido y ovoide, con cromatina fina, nucléolo aparente, citoplasma abundante que están ubicadas en el centro de los lóbulos y a veces distribuidas en rosetas o mosaicos laberínticos. Se observan linfocitos en cantidad variable, los cuales se disponen entre las células oscuras y claras o dentro del estroma fibroso que rodea al tumor. A veces hay depósito intercelular de material de membrana basal en un patrón en rompecabezas, siendo similar al observado en los cilindromas, dando lugar a formar mixtas conocidas como espiradenocilindromas. La resección quirúrgica es el tratamiento de elección. Es rara la recidiva del tumor benigno, contrario a lo observado en casos de espiroadenomas malignos en los cuales es frecuente.
Bibliografía
-
Martínez V, Aguilera V. Espiradenoma ecrino vascular. Rev Cent Dermatol Pascua .Vol. 12, Núm. 1. Ene-Abr 2003. Free text link
-
Tran K, DeFelice T, Robinson M, et al. Spiradenomas. Dermatol Online J. 2012;18(12):15. Free text link
CASO 45 *
16 de Noviembre
Dra. Carolina Cuéllar. Residente de Dermatopatología. Universidad CES. Medellín, Colombia.
Hombre anciano con placa verrugosa en piel de región lumbar. Diagnóstico clínico queratosis seborreica.
El diagnóstico más probable es:
a. Poroqueratosis
b. Queratosis seborreica
c. Carcinoma escamocelular in situ (fenómeno de Borst-Jadassohn)
d. Enfermedad de Paget extramamaria
e. Melanoma in situ
Respuesta 5 de diciembre 2016
 |  |  |
|---|---|---|
 |  |  |
 |  |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
Ca. escamocelular in situ (Borst-Jadassohn) Maria Janeth Vargas
Poroqueratosis Jairo Mesa Cock
Ca. escamocelular in situ (Borst-Jadassohn) Yamile Corredoira S
Ca. escamocelular in situ (Borst-Jadassohn) Carolina Delgado
Ca. escamocelular in situ (Borst-Jadassohn) Juan Carlos Garcés
Ca. escamocelular in situ (Borst-Jadassohn) Gabriel Casas
Ca. escamocelular in situ (Borst-Jadassohn) Gerardo Prada
Ca. escamocelular in situ (Borst-Jadassohn) Alexandra Maza
Ca. escamocelular in situ (Borst-Jadassohn) Anónimo
Ca. escamocelular in situ (Borst-Jadassohn) Marcela Saeb Lima
Ca. escamocelular in situ (Borst-Jadassohn) Carmen López
Ca. escamocelular in situ (Borst-Jadassohn) Elizabeth Ball
Ca. escamocelular in situ (Borst-Jadassohn) Hari Martínez Rivas
Ca. escamocelular in situ (Borst-Jadassohn) Enrique Loaiza
Ca. escamocelular in situ (Borst-Jadassohn) Clara Jaramillo
Ca. escamocelular in situ (Borst-Jadassohn) Esther Mariela Estrada
Ca. escamocelular in situ (Borst-Jadassohn) Juan Esteban Arroyave
Ca. escamocelular in situ (Borst-Jadassohn) Pablo José Erráez
Ca. escamocelular in situ (Fenómeno de Borst-Jadassohn)
El carcinoma escamocelular es el segundo cáncer de piel no melanoma más común en el mundo. Es importante conocer sus diversas formas de presentación histopatológica, incluyendo el epitelioma intraepidérmico de Borst-Jadassohn (1).
Clínicamente, esta variante se presenta como placas planas o verrugosas, marrones, en las extremidades inferiores, e histológicamente se considera un carcinoma escamocelular in situ (2).
El fenómeno de Borst-Jadassohn que se evidencia en este caso, es un hallazgo histopatológico común a varias patologías benignas y malignas, como queratosis seborreica, queratosis actínica, la enfermedad de Paget, algunos tumores ecrinos y el melanoma (2).
En 1904, Borst notó algunos nidos de células malignas en un carcinoma del labio, claramente separados del resto de la epidermis, lo cual interpretó como un crecimiento intraepidérmico del mismo carcinoma (2). Jadassohn en 1926 describió los nidos epidérmicos en ausencia de invasión dérmica, pensando que eran carcinomas basocelulares intraepidérmicos.
Fueron Mehregan y Pinkus quienes determinaron que Borst había descrito un carcinoma escamocelular del labio con invasión secundaria de la epidermis, y que lo que Jadassohn propuso como un carcinoma basocelular intraepitelial en realidad no lo era, y tenía una etiología mixta, por lo cual se considera que fue él quien identificó realmente este fenómeno histopatológico (2).
Bibliografia
1. Cassarino DS, DeRienzo DP, Barr DJ. Cutaneous squamous cell carcinoma: A comprehensive clonicopathologic classification. J Cutan Pathol 2006;33:191-206. Pubmed
2. Madke B, Doshi B, Pande S, Khopkar U. Phenomena in dermatology. Indian J Dermatol Venereol Leprol, 2011;77:264-75. Free text link
CASO 44
1 de Noviembre
Dra. Alejandra Ávila. Residente de Dermatología. Universidad Pontificia Bolivariana. Medellín, Colombia.
Mujer, 32 años, cuadro clínico de 15 años de evolución de edema de labio superior, asintomático, recurrente y episódico. Precedido de parestesias en mejilla derecha. Desde hace 5 años con edema orofacial continuo no doloroso. Sin antecedentes personales de importancia.
El diagnóstico más probable es:
1) Angioedema hereditario
2) Histiocitosis Intralinfática
3) Sarcoidosis
4) Síndrome de Melkersson-Rosenthal
5) Enfermedad de Crohn oral
Respuesta 15 de noviembre 2016
 |  |  |
|---|---|---|
 |  |  |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
S. Melkersson-Rosenthal María J. Vargas
S. Melkersson-Rosenthal Carmen López
S. Melkersson-Rosenthal Juan E. Arroyave
S. Melkersson-Rosenthal Juan C. Garcés
S. Melkersson-Rosenthal Gabriel Casas
S. Melkersson-Rosenthal Jairo Mesa Cock
S. Melkersson-Rosenthal Montse Molgo
S. Melkersson-Rosenthal Carolina Delgado
S. Melkersson-Rosenthal Gonzalo de Toro
S. Melkersson-Rosenthal Jesús Pérez G.
S. Melkersson-Rosenthal Elizabeth Ball
Queilitis granulomatosa (S. Melkersson-Rosenthal) Clara Jaramillo
S. Melkersson-Rosenthal Marcela Saeb
S. Melkersson-Rosenthal Enrique Loaiza
S. Melkersson-Rosenthal Maru Mazzei
S. Melkersson-Rosenthal Julia I Mesa
S. Melkersson-Rosenthal Yamile Corredoira S.
S. Melkersson-Rosenthal Anónimo
S. Melkersson-Rosenthal Juan David Mesa
S. Melkersson-Rosenthal Hari Martínez
Síndrome de Melkersson-Rosenthal
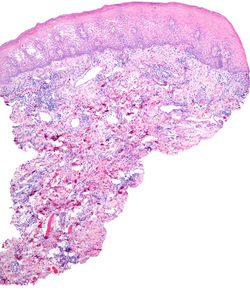
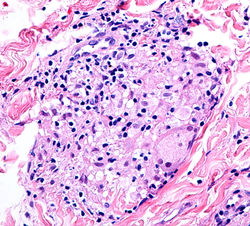
El síndrome de Melkersson-Rosenthal (SMR) es un trastorno neuro-mucocutáneo infrecuente, de curso recurrente y etiología desconocida, tiene una incidencia aproximada de 0,3 casos por 100.00 habitantes/año 1. Su presentación clínica se describe con una tríada que está presente de forma completa en el 25% de los casos e incluye: edema orofacial, parálisis facial recidivante y lengua fisurada (plicada o escrotal), las formas clínicas más prevalentes son mono y oligosintomáticas, siendo el edema de los labios el hallazgo principal (F1). Dicha manifestación se atribuye a la interrupción del flujo linfático y vascular por la presencia de granulomas 2,3. El SMR se ha relacionado con factores genéticos, reacciones alérgicas, infecciones, alteraciones del sistema inmune y finalmente, condiciones como la sarcoidosis y la enfermedad de Crohn 4.
La histopatología inicial se caracteriza por agregados de histiocitos, linfocitos y células plasmáticas de predominio perivascular asociado a edema inespecífico (F2 a 4). Cuando el SMR se ha establecido se encuentran granulomas no caseificantes en dermis, que pueden formarse y desvanecerse en días o semanas por lo que su ausencia no excluye el diagnóstico 5 (F5 y 6).
Desde el punto de vista clínico debe diferenciarse del edema angioneurótico, el angioedema hereditario, el síndrome de Ascher, la erisipela, neoplasias de las glándulas salivales, entre otros, desde el aspecto histológico, los granulomas del SMR son indistinguibles de los encontrados en la sarcoidosis (Ver caso 41 16 septiembre 2016) y la enfermedad de Crohn 5.
Referencias
1. Feng S, Yin J, Li et al. Melkersson-Rosenthal syndrome: a retrospective study of 44 patients. Acta Otolaryngol. 2014 Sep;134(9):977-81. PubMed
2. Saini AG, Sankhyan N, et al. Recurrent Facial Palsy and Electrophysiological Findings in Oligosymptomatic Melkersson Rosenthal Syndrome. Indian J Pediatr. 2016 Oct;83(10):1188-90. PuMed
3. Elias MK, Mateen FJ, Weiler CR. The Melkersson-Rosenthal syndrome: a retrospective study of biopsied cases. J Neurol. 2013 Jan;260(1):138-43. PubMed
4. Teresa Martínez-Menchón, Laura Mahiques. Síndrome de Melkersson-Rosenthal, Actas Dermo-Sifiliográficas, 94, (4), 2003, Pages 180-183. Free Text Link
5. Al-Hamad A. Et al. Orofacial Granulomatosis. Dermatol Clin. 2015 Jul;33(3):433-46. PubMed
CASO 43
16 de octubre 2016
Dra. Lina M. Aguirre. Residente de Dermatología. Universidad Pontificia Bolivariana. Medellín, Colombia.
Mujer joven con pequeña lesión nodular en cara. El diagnóstico más probable es:
-
Nevus intradermico
-
Tricoepitelioma
-
Linfadenoma cutáneo
-
Espiradenoma ecrino
-
Hidradenoma nodular
Respuesta 1 de noviembre 2016
 F1 |  F2 |  F3 |
|---|---|---|
 F4 |  F5 |  F6 |
 F7 |  F8 |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDO
Linfadenoma María janeth Vargas
Linfadenoma Anónimo
Linfadenoma Patrick Agostini
Linfadenoma Clara Jaramillo
Linfadenoma Angela Seidel
Linfadenoma Alexandra Maza
Tricoepitelioma Gloria Mendoza de S
Linfadenoma Julia Mesa
Linfadenoma Elizabeth Ball
Linfadenoma Gabriel Varela
Linfadenoma Juan E. Arroyave
Lindadenoma Jairo Mesa Cock
Linfadenoma Verónica Posso
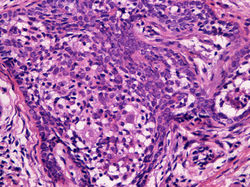
Linfadenoma cutáneo
Neoplasia epitelial benigna poco frecuente, descrita por Santa Cruz y Barr en 1987, el cual ha recibido diferentes nombres; tricoblastoma adamantinoide linfotrópico, y actualmente linfadenoma cutáneo (LC), considerado una variante del tricoblastoma nodular 1. Aunque este tumor es de comportamiento benigno, es semejante clínica e histológicamente al carcinoma basocelular (CBC).
Afecta a personas de todas las edades, principalmente adultos de sexo masculino. La localización mas frecuente es en la cabeza y el cuello. Se presenta clínicamente como una pápula o nódulo solitario de 1 a 2.5 cm (F1), no ulcerado, asintomático y de crecimiento lento 2.
La histología del tumor, se caracterizada por una lesión nodular intradérmica (F2), no encapsulada, bien circunscrita , con nidos o cordones trabeculares de células epiteliales basaloides de núcleo hipercromático (F3, F4), con una o dos capas en empalizada periférica, similar al CBC (F5, F6). Las células tumorales en el centro de la lesión son de citoplasma claro abundante, también hay presencia de infiltrado linfocitario e histiocitario dentro del tumor y en el estroma circundante (F7, F8). Pueden tener conexión con la epidermis o folículos pilosos 1,2. Usualmente no presentan actividad mitótica, invasión perineural ni intravascular.
La inmunohistoquímica puede ayudar a establecer el diagnóstico, destacando el patrón de tinción de la citoqueratina 17, el cual es difuso en los nidos basaloides del CBC, mientras que en el LC el patrón es incompleto en el borde y en parches en el centro de los nidos 2.
Referencias
1. Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014 Jun 3;6(2):5358. Free Text Link.
2. Goyal A, Solus JF, Chan MP, et al. Cytokeratin 17 is highly sensitive in discriminating cutaneous lymphadenoma (a distinct trichoblastoma variant) from basal cell carcinoma. J Cutan Pathol. 2016 May;43(5):422-9. PubMed
CASO 42*
3 de octubre 2016
Dra. Carolina Barrera. Residente de Dermatopatología. Universidad CES. Medellín, Colombia.
Mujer de 30 años con abundante cabellera. Se queja de sensación de arena al peinarse. El diagnóstico más probable es:
-
Tricomicosis
-
Pediculosis
-
Piedra blanca
-
Molde peripilar
-
Tricorrexis nodosa
Respuesta 16 de octubre 2016
 |  |  |
|---|---|---|
 |  Luz Polarizada |  |
 |  HE |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
Piedra blanca Natalia Valderrama
Piedra blanca Angela Seidel
Piedra blanca Gerardo Prada
Molde peripilar Marcelo Toro
Piedra blanca Clara Jaramillo
Piedra blanca Juan E. Arroyave
Piedra blanca Luisa F Rios
Molde peripilar Elizabeth Ball
Piedra blanca Patrick Agostini
Piedra blanca Jairo Mesa Cock
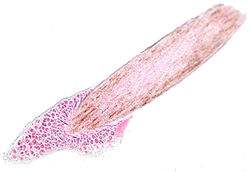
PIEDRA BLANCA
Fue descrita por primera vez por Beigel en 1865. La piedra, es una infección fúngica del eje del pelo. Dos variedades de piedra se pueden ver, uno llamado la piedra blanca y otro la piedra negra.
La piedra blanca es causada por Trichosporon beigelii, también conocida como tiña nodosa, tricosporonosis nodosa y tricomicosis nodular. Se encuentra en los países semitropicales y templados. La piedra negra es generalmente causada por el hongo Piedrae hortae y Trichosporon ovoides, se encuentra sobre todo en los países tropicales como Suramérica y el Sudeste de Asia.
La piedra blanca se caracteriza clínicamente por nódulos de color marrón claro que pueden rodear todo el eje del pelo, estos son suaves y la masa de hongos pueden ser fácilmente separados del cabello. Se confirma el diagnóstico con hallazgos microscópicos observándose micelios que se fragmentan en artrosporas rectangulares y blastoconidios. El diagnóstico diferencial es muy importante ya que, puede pasar desapercibido o confundirse con la pediculosis.
La terapia clásica y más eficaz para la piedra blanca ha sido y sigue siendo el corte y afeitado de los pelos. También se recomiendan anti fúngicos tópicos.
Bibliografía
-
Saxena S, Uniyal V, Bhatt RP. Inhibitor effect of essential oils against Trichosporon ovoides causing Piedra hair infection. Braz J Microbiol. 2012 Oct;43(4):1347-54. Free text link
-
Tamble SA, Dhurat SR, Kumar CA, Thakare P, Lade N. Two cases of scalp white piedra caused by Trichosporon ovoides. Indian J Dermatol Venereol Leprol. 2009 May-Jun;75(3):293-5. Free text link
CASO 41
16 de septiembre 2016
Dr. Andrés Flórez Profesor de Dermatopatología. Universidad CES. Medellín, Colombia.
Hombre de 52 años con nódulo sobre cicatriz en region tenar derecha. Historia de trauma. El diagnóstico más probable es:
-
Neurofibroma
-
Sarcoma sinovial
-
Fibromixoma acral
-
Sarcoidosis
Respuesta 30 de septiembre 2016
 |  |  |
|---|---|---|
 |  Luz polarizada |  |
 |  Retículo |  Retículo |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
Sarcoidosis Marcelo Toro
Sarcoidosis Esther Mariela Estrada
Sarcoidosis Gerardo Prada
Sarcoidosis Julia Mesa
Neurofibroma Anónimo
Sarcoidosis Camila Montoya
Sarcoidosis Iva Johansson
Sarcoidosis María Janeth Vargas
Sarcoidosis Patrick Agostini
Sarcoidosis in a scar and foreign body reaction Jaime Arturo Mejía
Sarcoidosis Jairo Mesa Cock
Sarcoidosis Carlos Cortés Caballero
Sarcoidosis Rita María Paéz
Sarcoidosis Maria Gloria Mendoza
Sarcoidosis Nathalie Lascano
Reacción sarcoidal Ricardo Rueda
Sarcoidosis Juan M. González
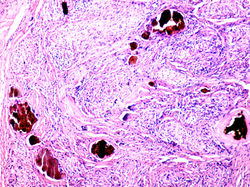
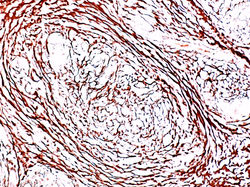
Sarcoidosis
Es una enfermedad multisistémica de etiología desconocida caracterizada por la presencia en los órganos afectados de granulomas sarcoideos. Suele afectar a jóvenes y adultos de mediana edad y con frecuencia se presenta con adenopatías hiliares bilaterales, infiltrados pulmonares y lesiones oculares y cutáneas. El diagnóstico se establece con los hallazgos clinicorradiológicos adecuados y deben excluirse los granulomas de causas conocidas y las reacciones sarcoides locales. En cuanto a la sarcoidosis cutánea tenemos que estas lesiones cutáneas se clasifican en específicas y no específicas. Las lesiones específicas son aquellas que histológicamente presentan granulomas sarcoides. Las lesiones inespecíficas son aquellas lesiones observadas con mayor frecuencia en pacientes portadores de una sarcoidosis sistémica pero que también pueden observarse aisladas o asociadas a otras enfermedades y cuya biopsia no permite el diagnóstico de la enfermedad. Las lesiones específicas más frecuentes son maculopápulas, placas, lupus pernio, sarcoidosis de las cicatrices y sarcoidosis subcutánea, y la lesión no específica más importante es el eritema nodoso. La lesiones “específicas” presentan el mismo patrón histológico que es la presencia de granulomas tuberculoides no necrotizantes, redondeados, diseminados, los cuales están constituidos por abundantes histiocitos epitelioides, número variable de células gigantes multinucleadas con escasa o nula corona linfocitaria (fig.1,2,3,7). En las células gigantes pueden evidenciarse inclusiones no especificas tales como cuerpos asteroides y de Schaumann o partículas cristalinas, por lo que debe someter la preparación a la luz polarizada, siendo refringentes o no refringentes (fig.5). Con la técnica de reticulina (fig.8,9) se revela una red de fibras reticulares rodeando y penetrando las células epitelioides, a diferencia de los granulomas infecciosos en los que la reticulina permanece periférica.
No olvidar que el diagnostico de sarcoidosis siempre es basado en la exclusión de otras enfermedades infecciosas y no infecciosas que se presentan con granulomas.
Bibliografía
-
Gatti F., Prahl P., Troielli P. et al. Sarcoidosis. Un enfoque global. Med Cutan Iber Lat Am 2008;36(4):165-182.
-
Fortuño Y., Gallego I., Marcoval J. Sarcoidosis cutánea. Actas Dermosifiliogr 2004;95:137-53 - Vol. 95 Núm.3.
CASO 40
1 de septiembre 2016
Dra. Catalina Cuéllar. Residente Dermatopatología. Universidad CES. Medellín, Colombia.
Mujer de 40 años con masa subescapular derecha. Trabaja en un textil artesanal. El diagnóstico más probable es:
-
Elastofibroma
-
Hibernoma
-
Tumor fibroso solitario
-
Hamartoma fibroso
Respuesta 15 de septiembre 2016
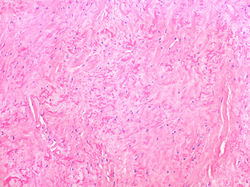 F1 | 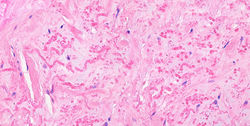 F2 |  F3 |
|---|---|---|
 F4 Elástico |  F5 Elástico | 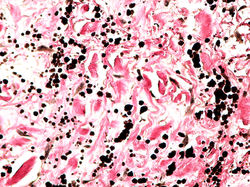 F6 Elástico |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
Elastofibroma Víctor Delgado
Elastofibroma María Janeth Vargas
Elastofibroma Camila Montoya
Elastofibroma Julia Mesa
Elastofibroma Esther Mariela Estrada
Elastofibroma Katty Lacayo
Elastofibroma Verónica Posso
Elastofibroma Gerardo Prada
Elastofibroma dorsi Gabriel Varela
Elastofibroma Anónimo
Elastofibroma Hari Martínez
Elastofibroma (dorsi) Clara Jaramillo
Elastofibroma Jairo Mesa Cock
Elastofibroma Rita María Paéz
Elastofibroma Carlos Cortés Caballero
Elastofibroma
El elastofibroma es un tumor de tejidos blandos infrecuente, de crecimiento lento, que se presenta en la región infraescapular, en mujeres de edad avanzada (1). Usualmente es asintomático, pero se puede asociar a dolor, edema, o limitación del movimiento del hombro; puede ser único o bilateral (1).
La histogénesis de este tumor es controvertida, ya que algunos lo consideran una neoplasia, pero también existe la teoría de que se trata de una hiperproliferación del tejido conectivo debido a la fricción de la escápula y la pared torácica por el trauma repetitivo (1,2,3). La fricción induce una producción aumentada de tejido conectivo, con degeneración de las fibras de colágeno; la cual a su vez se asocia con aumento en la cantidad de matriz elástica producida, alternando con el depósito de grasa hiperplásica en la lesión (2). Se ha propuesto también que puede originarse por insuficiencia vascular, que induce fibromatosis reactiva y degeneración elastótica (2).
La anatomía macroscópica del elastofibroma muestra una masa irregular, no encapsulada, de consistencia cauchosa, que al corte presenta un tejido fibroelástico blanquecino mezclado con tejido adiposo (1). La histopatología es característica, en la forma de un tumor dérmico, no encapsulado e hipocelular con haces fibrosos y numerosas fibras elásticas dispersas, asociados a un estroma mucoide y adipocitos maduros (F1) (1). Las fibras elásticas son grandes, intensamente eosinofílicas, con tendencia a fragmentarse en glóbulos o discos serrados típicos, que se disponen en filas, como un collar (F2, F3) (2). El tumor es positivo para vimentina, CD34 y lisozima y negativo para actina de músculo liso, S-100, desmina y p53 (1,3) La tinción elástica de Verhoeff es positiva (F4-F6)(1).
El cuadro clínico y la imagen histológica son tan características que prácticamente no hay diagnósticos diferenciales (1,3).
Bibiografía
1. Patnayak R, Jena A, Settipalli S, et al. Elastofibroma: An uncommon tumor revisited. J Cutan Asthet Surg 2016;9:34-7. Free text link
2. Braham E, Hergli I, Boudaya MS, et al. Elastofibroma of scapula: a case report and literature review. Ann Transl Med 2013;1:31. Free text link
3. Di Vito A, Scali E, Ferraro G, et al. Elastofibroma dorsi: A histocheminal and immunohistochemical study. Eur J Histochem 2015;59:2459. Free text link
CASO 39
1 de agosto 2016
Dra. Luz Marina Gómez V. Profesora Programa Dermatología UPB. Medellín, Colombia..
Dr. Andrés Flórez Profesor de Dermatopatología. Universidad CES. Medellín, Colombia.
Mujer de 45 años con pigmentación facial. Con las imágenes suministradas el diagnóstico más probable es:
-
Melasma
-
Liquen pigmentógeno
-
Melanocitosis dérmica
-
Ocronosis exógena
Respuesta 15 de agosto 2016
 F1 |  F2 | 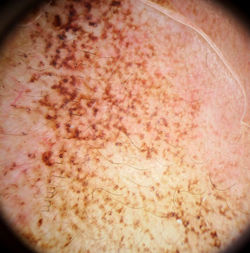 F3 |
|---|---|---|
 F4 |  F4 |  F6 |
 F7 |  F8 |  F9 Rudolph Virchowhttps://commons.wikimedia.org/wiki/File%3ARudolf_Virchow_NLM3.jpg |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
Ocronosis exógena María Janeth Vargas
Ocronosis exógena Anónimo
Ocronosis exógena Ernesto Peña
Ocronosis exógena Hari Martínez Rivas
Ocronosis Gerardo Prada
Ocronosis Gabriel Varela
Ocronosis exógena Liliana De Gracia
Ocronosis exógena Jaime Arturo Mejía
Ocronosis exógena Carmen López
Ocronosis Juan M. González
Ocronosis exógena Jairo Mesa Cock
Ocronosis exógena Ana Cristina Ruíz
Ocronosis exógena Patrick Agostini
Ocronosis exógena Elsa Bibiana Peña
Ocronosis exógena Juan Esteban Arroyave
Ocronosis exógena Luisa Fernanda Ríos
Ocronosis exógena Clara Jaramillo
Ocronosis exógena Julia Mesa
Ocronosis exógena Camila Montoya
Ocronosis exógena Elizabeth Ball de Picon
Ocronosis exógena Olga Mercedes Álvarez O.
Ocronosis exógena Katty Lacayo
Ocronosis exógena Esther Mariela Estrada
OCRONOSIS EXÓGENA (OE)
La ocronosis es una patología que puede ser de tipo endógeno o exógeno. La primera se trata de un trastorno autosómico recesivo por alteración del metabolismo del ácido homogentísico, conocido también como alcaptonuria. Fue descrita originalmente por Rudolph Virchow (F9) en la autopsia de un hombre de 67 años, el 8 de mayo de 1865. Su descripción apareció en la edición de octubre de 1866 en Vichows Archiv.1,2 Virchow pensó que la pigmentación observada estaba estrechamente relacionada con la acumulación de pigmente de lipocromo (pigmento de desgaste).
La OE fue descrita en 1912 por Beddard y Plumtre en un paciente que utilizaba fenol para el tratamiento de una úlcera en la pierna. El primer reporte relacionado con el uso de hidoquinona, como en este caso, lo realizo Findlay en 1975. La enfermedad también se ha observado como consecuencia del uso de medicamentos para la malaria, resorcinol y mercurio.
Clínicamente se observa hiperpigmentación color marrón/grisácea en forma de pápulas y parches con taponamiento folicular a la dermatoscopia, más frecuente en arcos cigomáticos y región supraciliar (F1-F3), aunque puede aparecer en cualquier zona donde se haya hecho la aplicación del producto desencadenante. Se piensa que la hiperpigmentación se debe a la inhibición de la enzima oxidasa homogentısico por la hidroquinona. 3
Los diagnósticos clínicos diferenciales principales son: el melasma (no se observa taponamiento folicular), aunque es frecuente observar hallazgos de melasma superpuestos. La hiperpigmentación por acumulación de metales (“pseudocronosis”) es generaliza y afecta las mucosas. 3
La histopatología es diagnóstica observando en la dermis superficial, cuerpos de color ocre que adoptan cualquier forma, pero más característicamente simulando un banano (F4-F8). No hay alteración de epidermis ni de anexos, aunque puede haber hallazgos asociados a foto exposición crónica tales como elastosis solar, telangiectasias y mínimo infiltrado inflamatorio. Prácticamente no hay diagnóstico diferencial desde el punto de vista histológico. 3
Bibiografía
1.Virchow, R. Archiv f. pathol. Anat. (1866) 37: 212.
2. Virchow RL. Rudolph Virchow on ochronosis.1866. Arthritis Rheum. 1966 Feb;9(1):66-71. Free text link
3. Charlín R, Barcaui CB, Kac BK, et al. Hydroquinone-induced exogenous ochronosis: a report of four cases and usefulness of dermoscopy. Int J Dermatol. 2008 Jan;47(1):19-23. Free text link
CASO 38
15 de julio 2016
Dra. María Catalina Cuéllar. Residente de Dermatopatología. Universidad CES. Medellín, Colombia.
Mujer de 50 años con lesón genital. Prurito intenso. Con las imágenes suministradas el diagnóstico más probable es:
-
Carcinoma escamocelular
-
Liquen plano
-
Psoriasis inversa
-
Líquen escleroso
Respuesta 31 de Julio 2016
 |  F1 |  F2 |
|---|---|---|
 F3 |  F4 |  F5 |
 F6 |  F7 |  F8 |
 F9 |  F10 |  F11 |
 F12 | 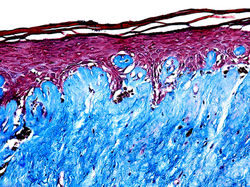 F13 Tricrómico |
Haga clic arriba ^ para vista de pantalla completa
RESPUESTAS Y COMENTARIOS RECIBIDOS
Liquen escleroso Olga Maria Oiticica Harris
Liquen escleroso y atrófico Juan Manuel González
Liquen escleroso. No siempre hay atrofia Jairo Mesa Cock
Liquen escleroso María Janeth Vargas
Liquen escleroso y atròfico Guillermo Ramos R.
Liquen escleroso Esther Mariela Estrada
Liquen escleroso Gerardo Prada
Liquen escleroso y atrófico Lili de Gracia
Liquen escleroso Carlos Cortés Caballero
Liquen escleroso Víctor Argueta Sandoval
Liquen escleroso y atrófico Gustavo Matute
Liquen escleroso Clara Jaramillo
Liquen escleroso María del Consuelo Gómez
Liquen escleroso y atrófico Olga Lucía castaño
Liquen escleroso y atrófico Fernando Brenner
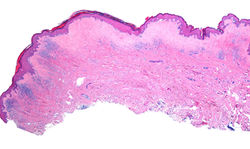
Líquen Escleroso (LE)
El LE es una enfermedad inflamatoria crónica mucocutánea que ocurre principalmente en mujeres postmenopáusicas. Las lesiones tienen aspecto de porcelana y se pueden acompañar de púrpura, esclerosis, fisuras, ampollas y un grado variable de cicatrización progresiva (1). La localización más frecuente es anogenital, pero también se puede presentar en el torso (1).
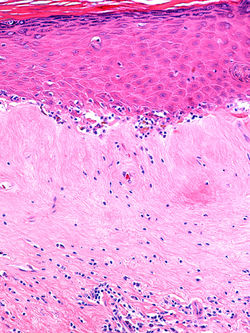
En la histopatología de las lesiones tempranas de LE se observa hiperqueratosis e hipergranulosis de los folículos pilosos, acantosis leve irregular y engrosamiento de la memberana basal (F1-F7) , con presencia de edema subepitelial, homogenización del colágeno (F8-F13) y telangiectasias bajo la membrana basal (F11, F12). Puede encontrarse un infiltrado inflamatorio linfocítico liquenoide o intersticial con exocitosis de linfocitos(F3, F11, F12) (2). Cuando la enfermedad progresa, se observa hiperqueratosis, atrofia epidérmica con aplanamiento de la red de crestas, daño vacuolar de la basal, pérdida de elásticas, hialinización de la dermis papilar y un infiltrado linfocítico subyacente. A veces puede haber separación artifactual de la epidermis de la dermis sin que esto represente una verdadera ampolla (F9, F10) (2).
En cuanto a los diagnósticos diferenciales, es importante descartar el carcinoma escamocelular, ya que 5% de los pacientes con LE pueden llegar a presentarlo. Hay queratinocitos con atipia proliferando desde la epidermis e invadiendo la dermis. El líquen plano se puede yuxtaponer al LE, pero se diferencia en que se presenta hiperqueratosis, acantosis, hipergranulosis, aspecto serrado de la red de crestas y un infiltrado linfocítico en banda que oculta la unión dermoepidérmica, con queratinocitos apoptósicos. La psoriasis inversa muestra hiperqueratosis, acantosis regular de la red de crestas, hipogranulosis, adelgazamiento suprapapilar y microabscesos de Munro.
Bibliografía
1. Kirtschig G. Lichen sclerosus – Presentation, diagnosis and management. Dtsch Artzebl Int 2016;113:337-43 Free text link
2. Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. An update. Am J Clin Dermatol 2013;14:27-47 F ree text link